

Mesenchymal and adrenergic cell lineage states in neuroblastoma possess distinct immunogenic phenotypes
- PMID: 36138189
- PMCID: PMC10171398
- DOI: 10.1038/s43018-022-00427-5
Mesenchymal and adrenergic cell lineage states in neuroblastoma possess distinct immunogenic phenotypes
Abstract
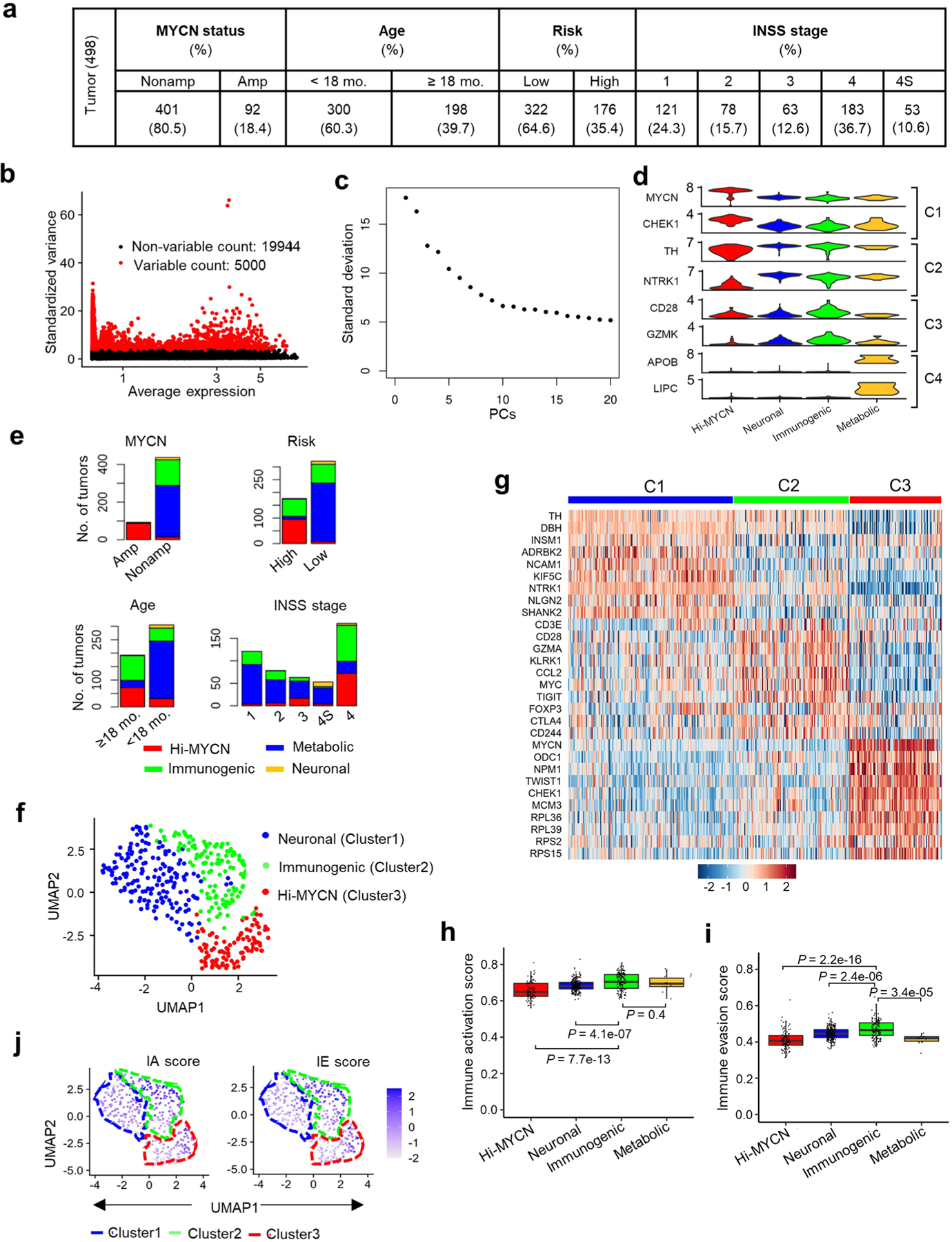
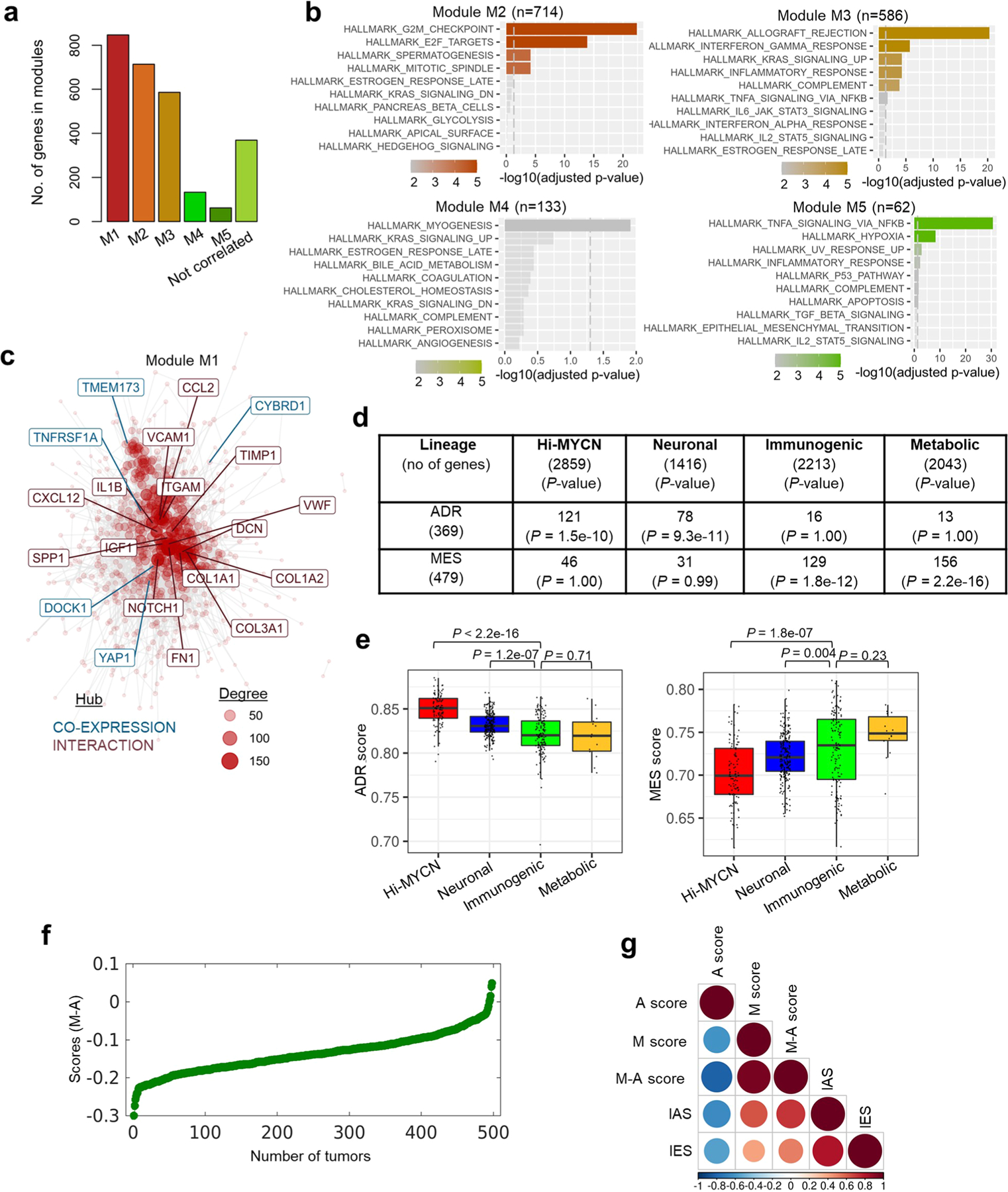
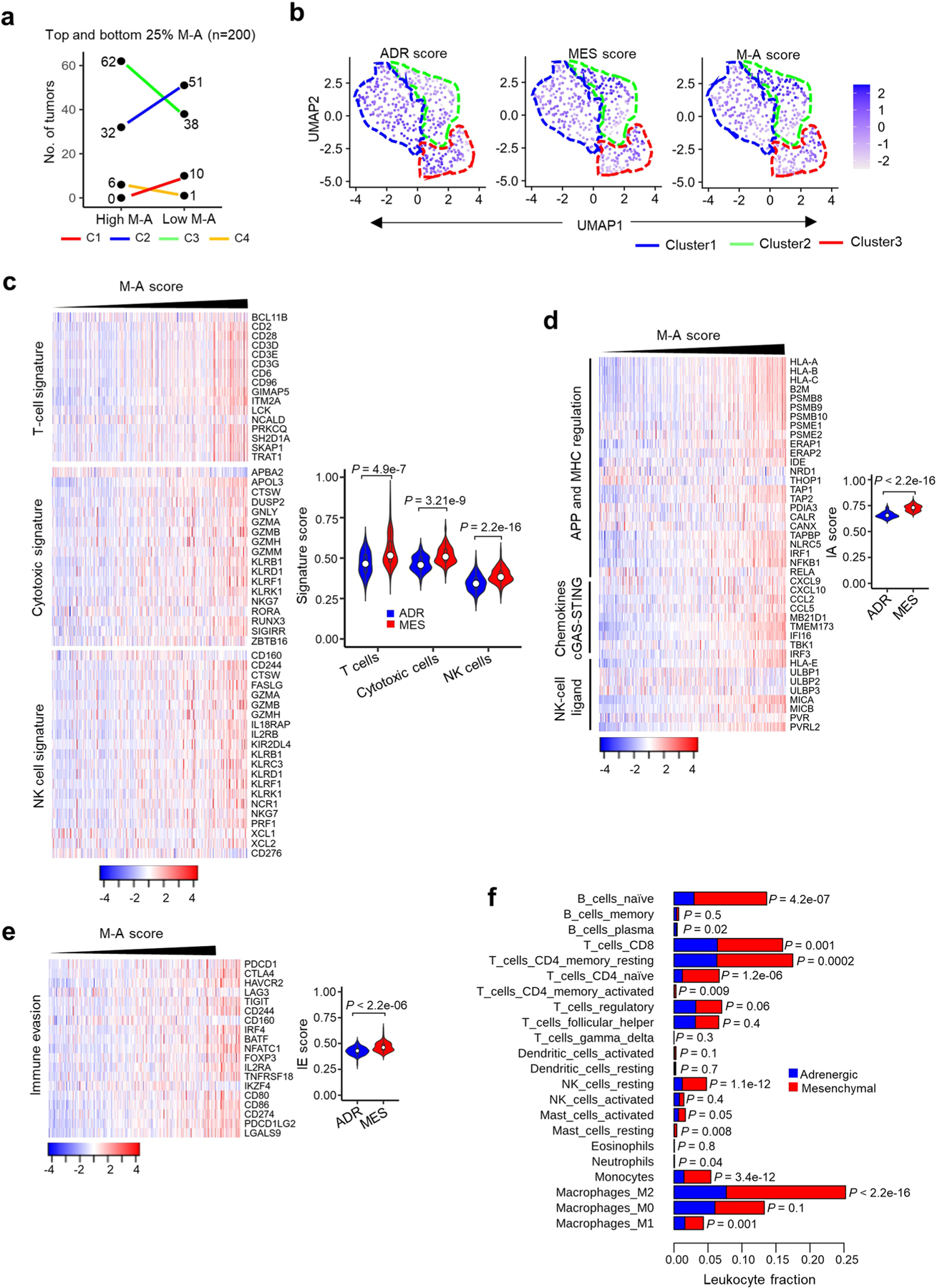
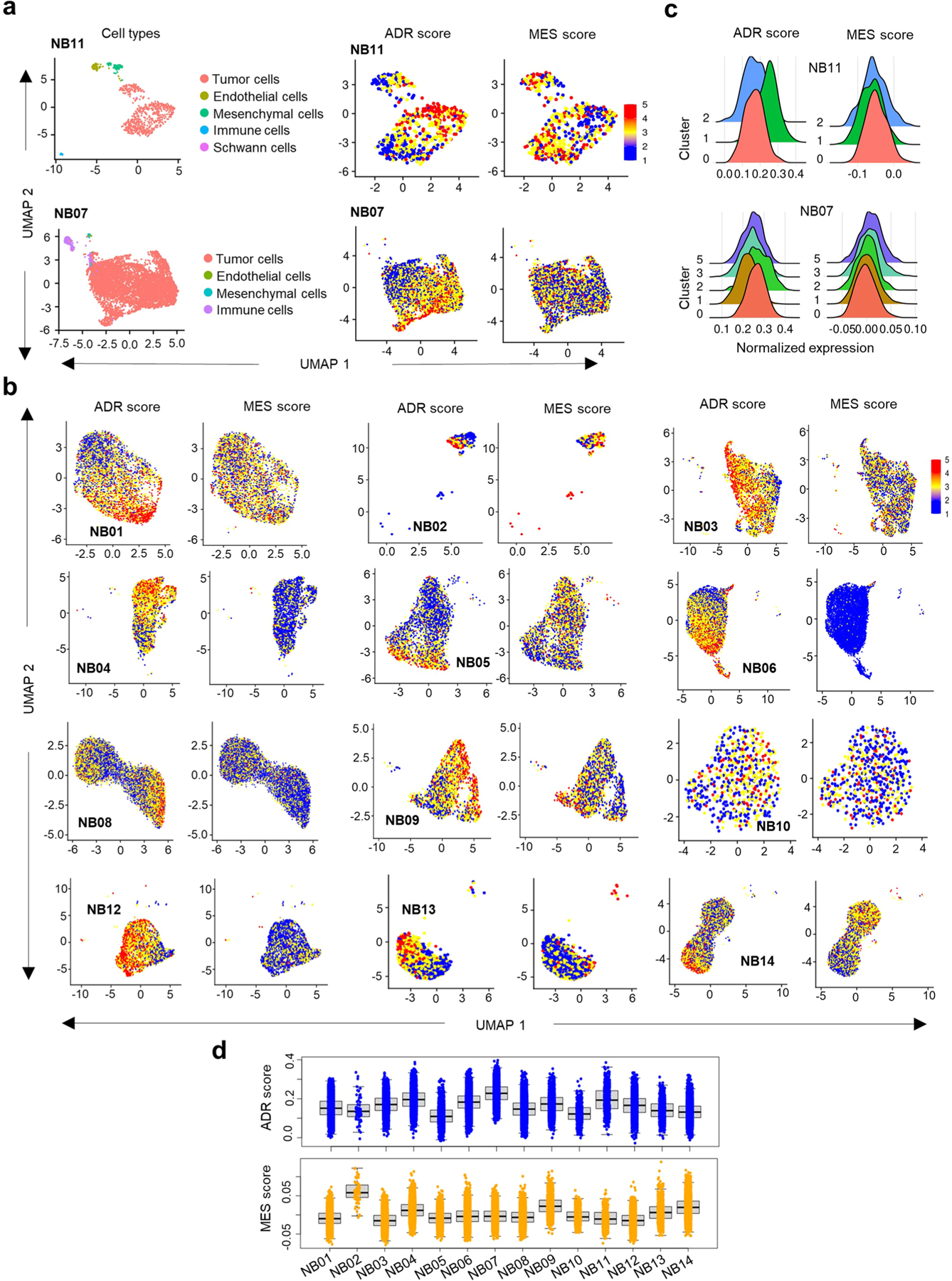
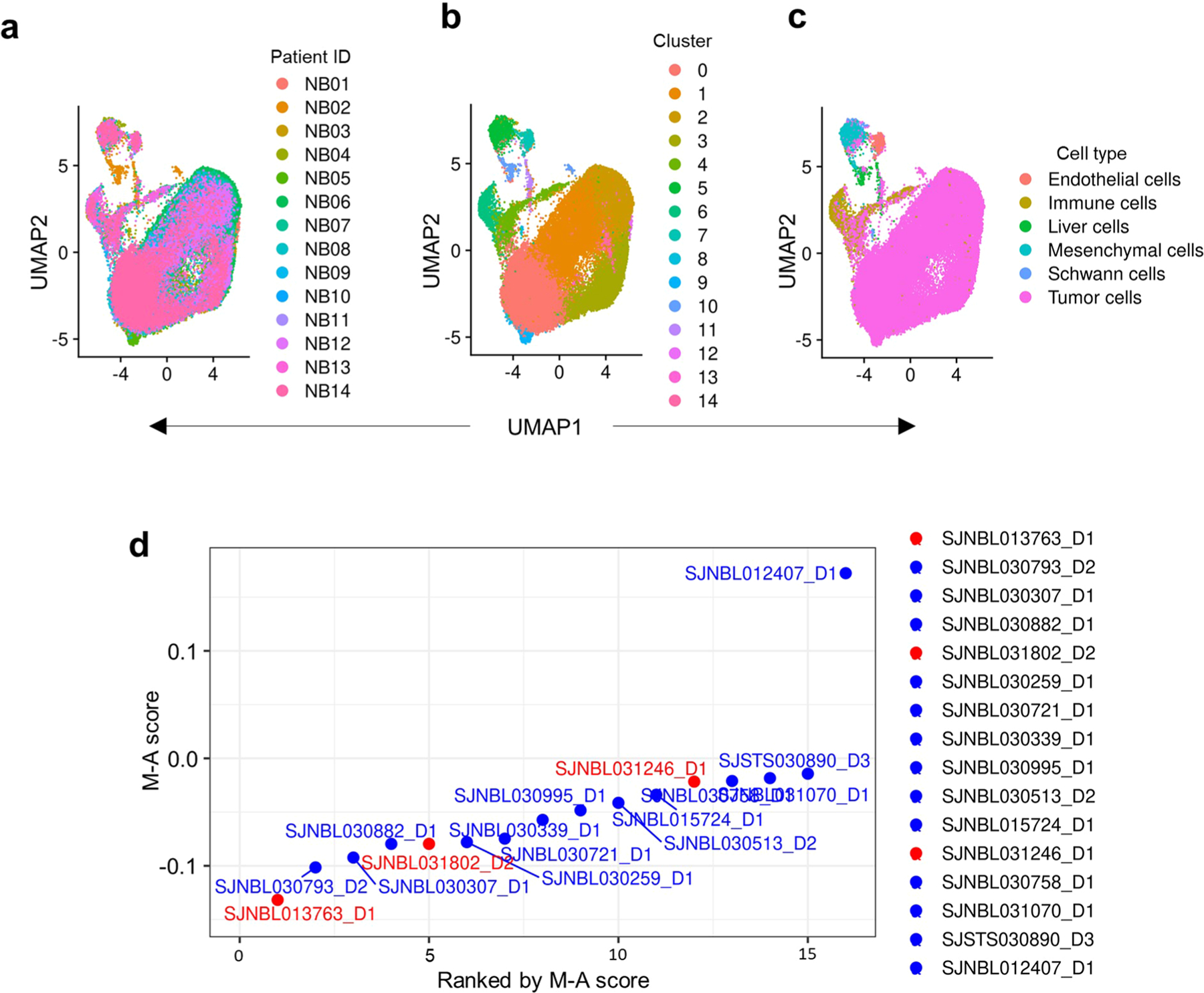
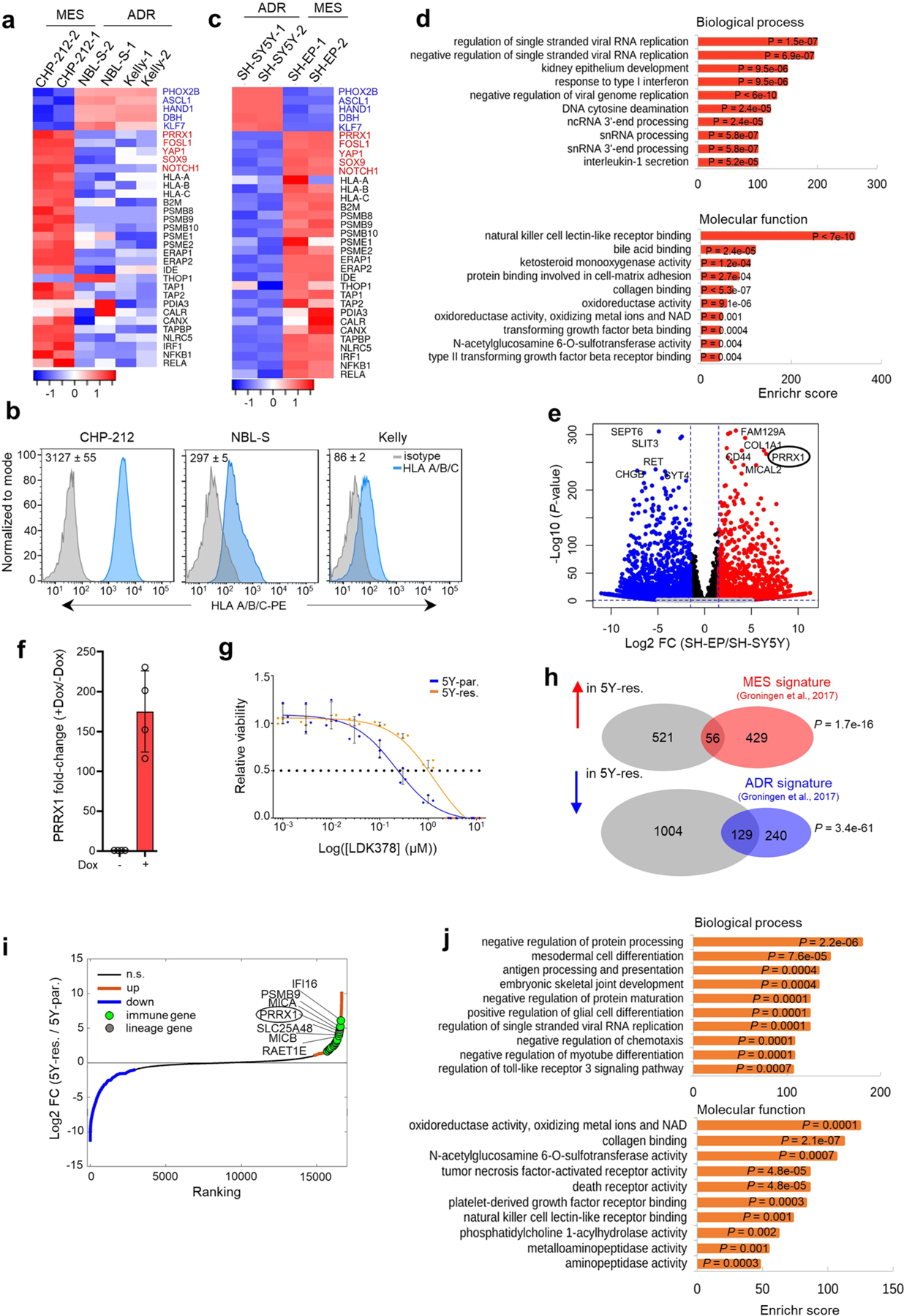
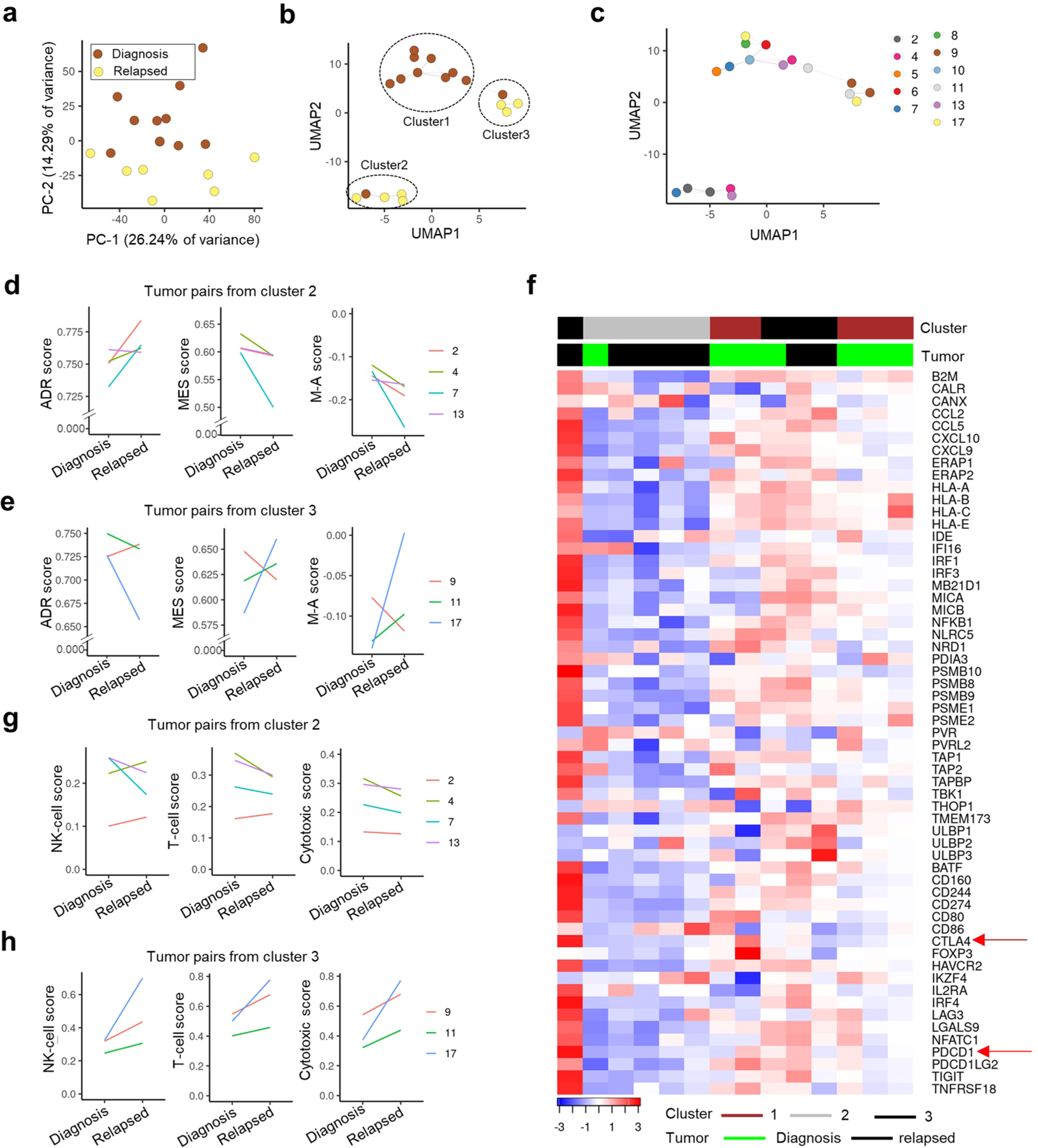
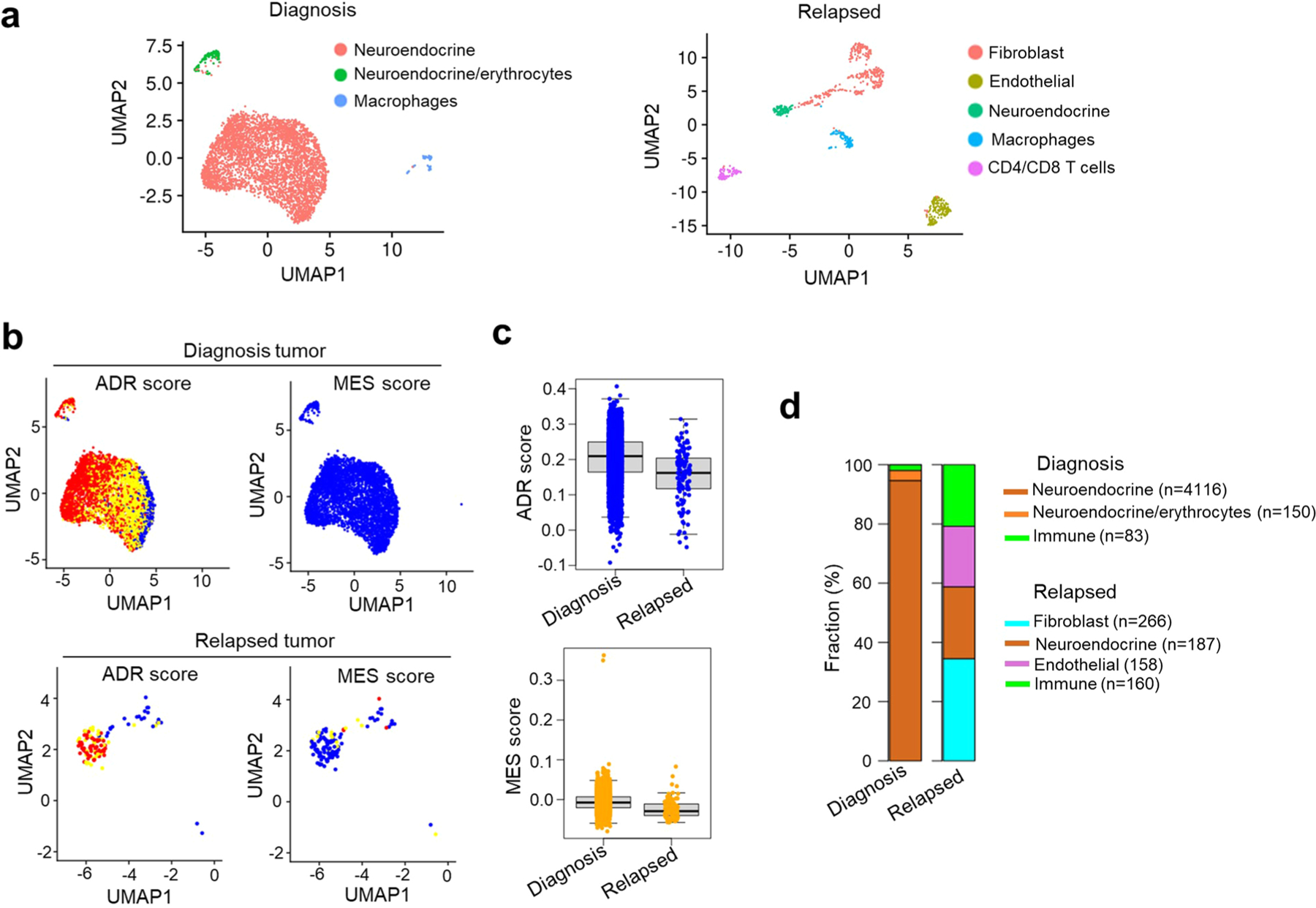
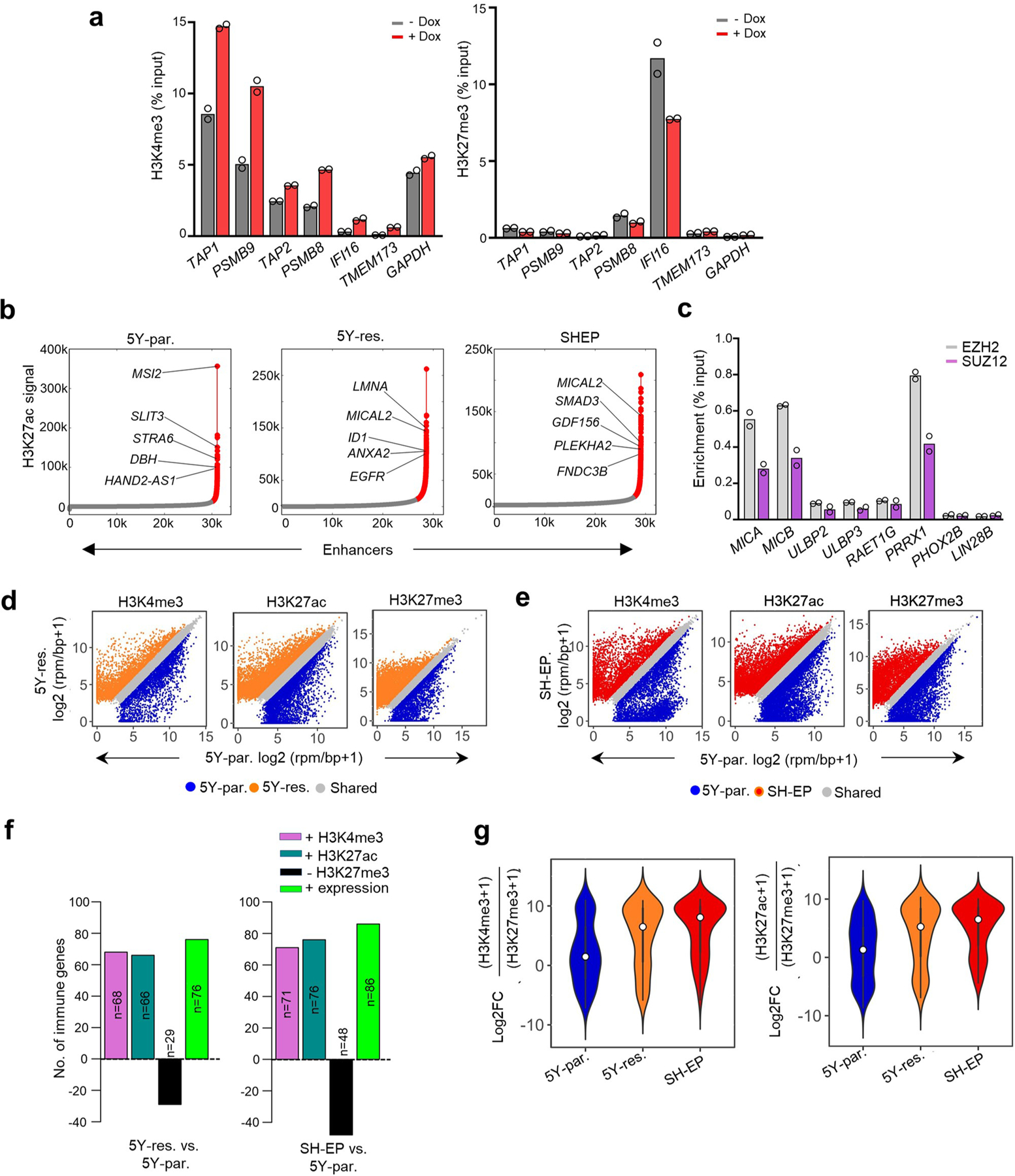
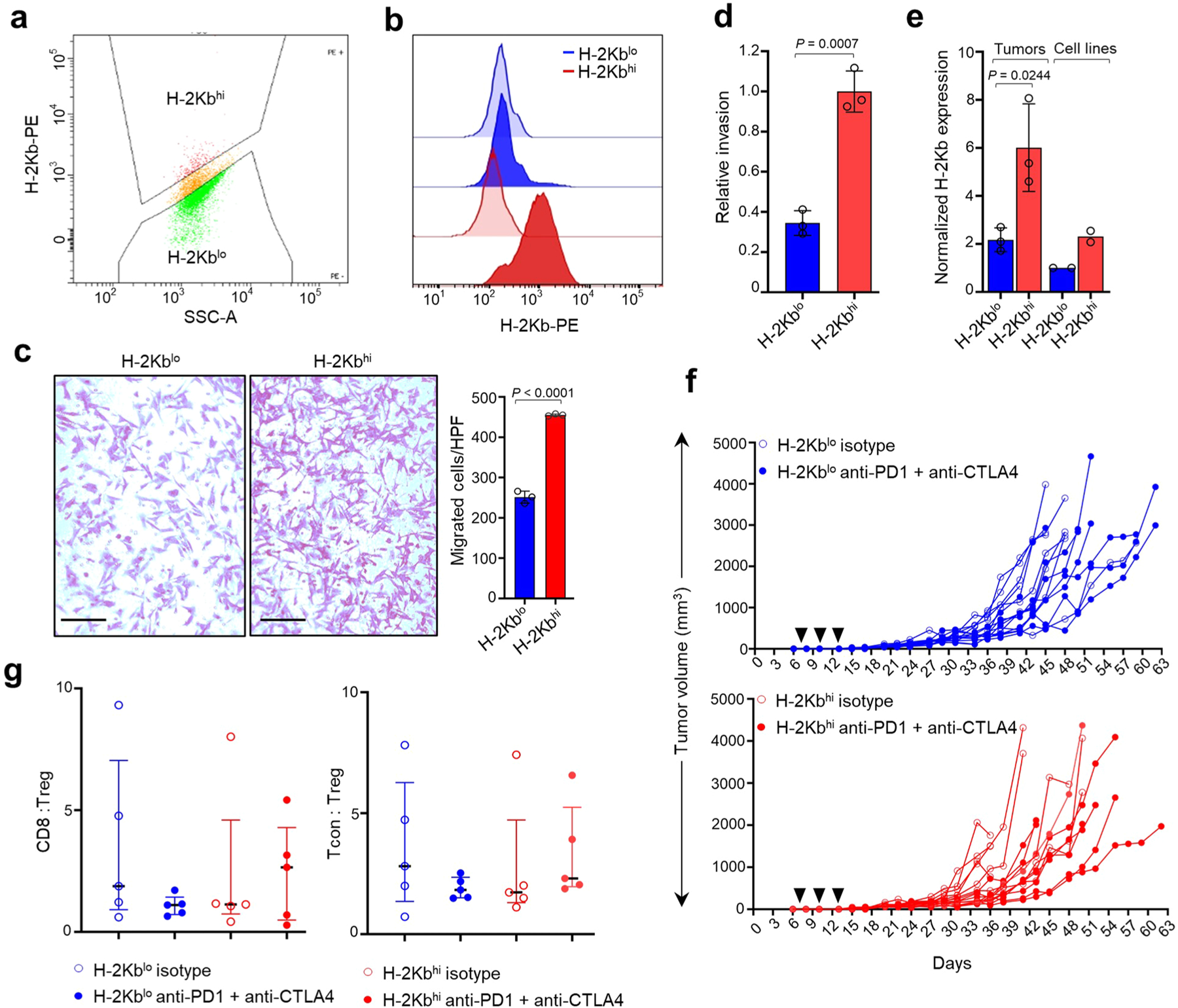
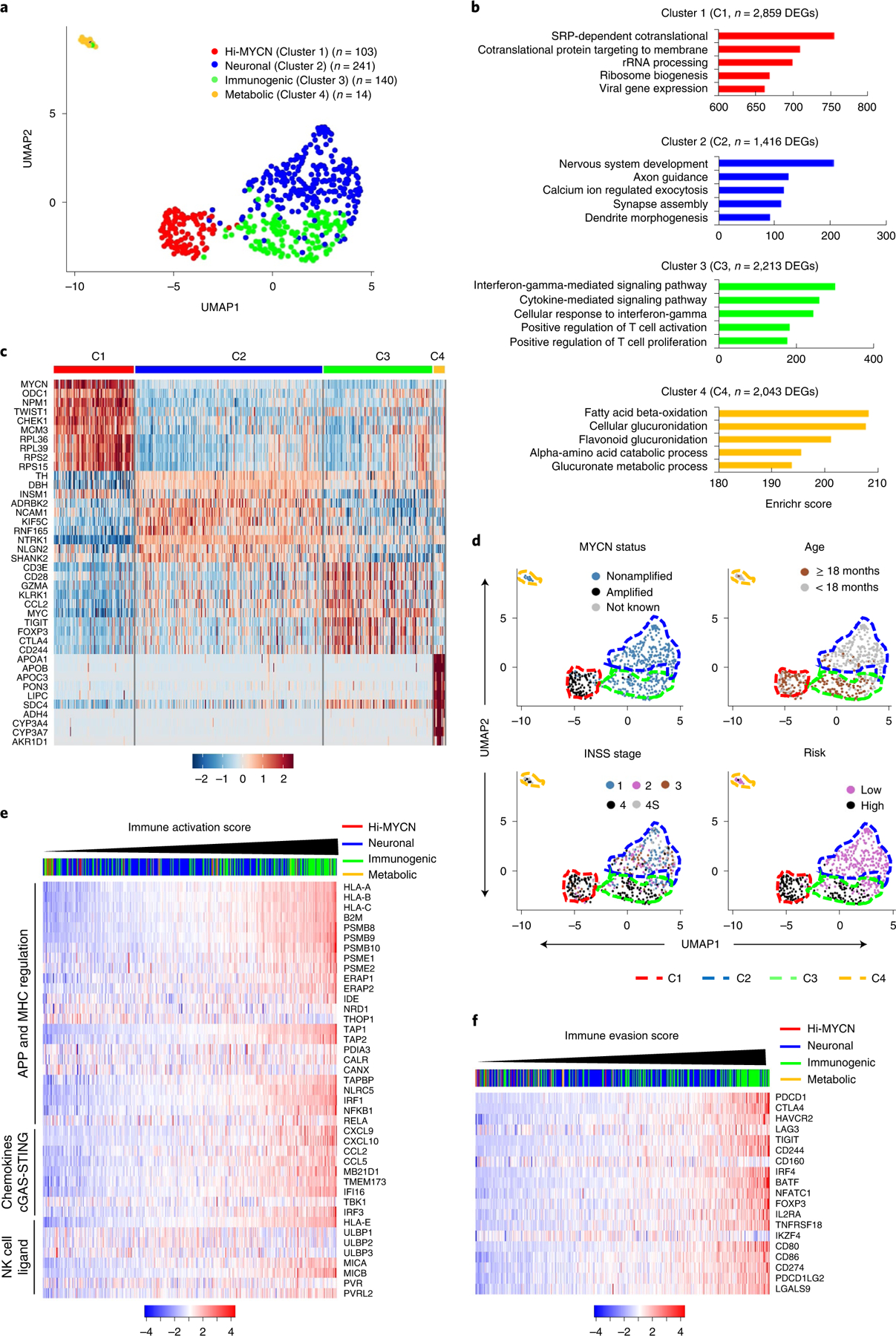
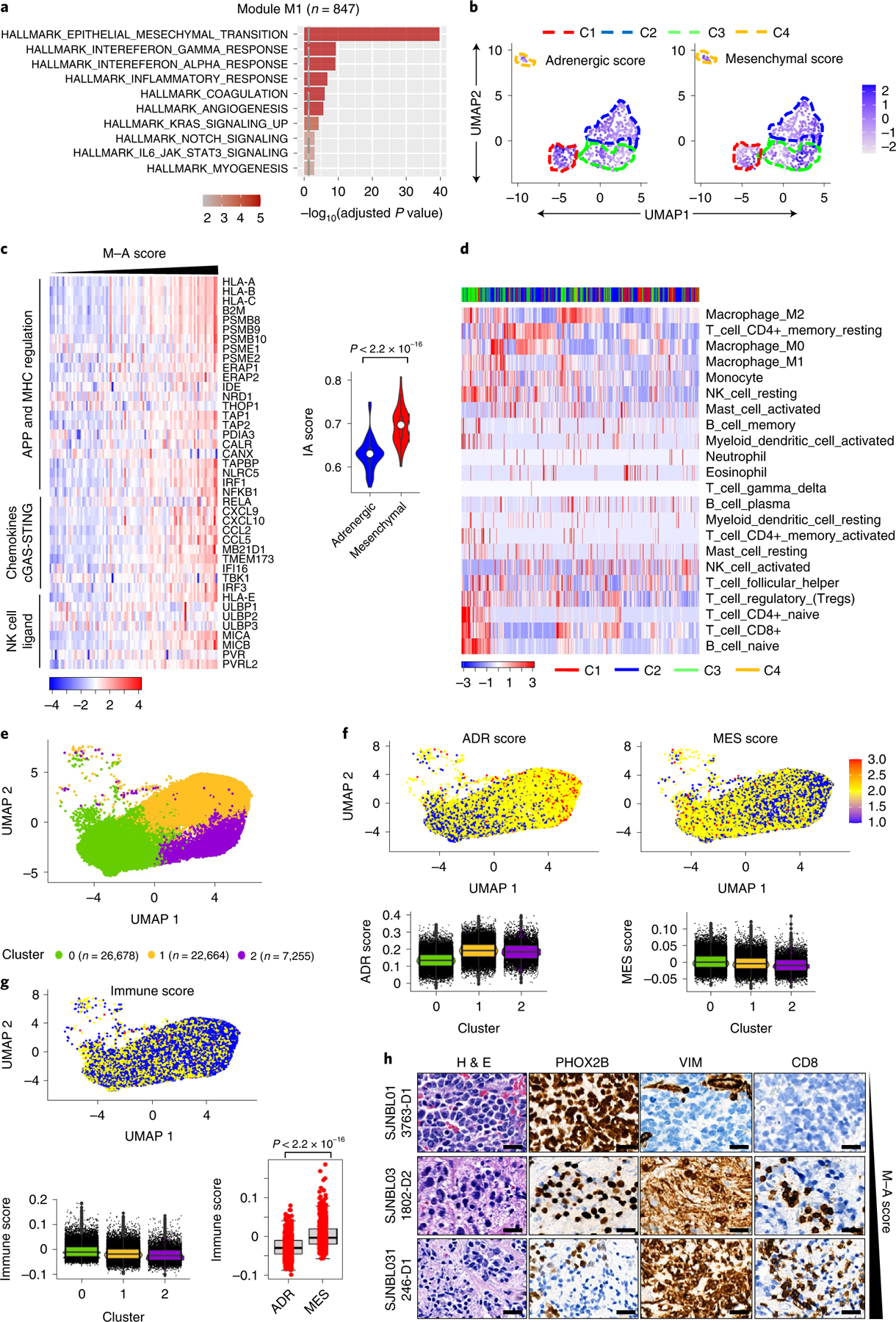
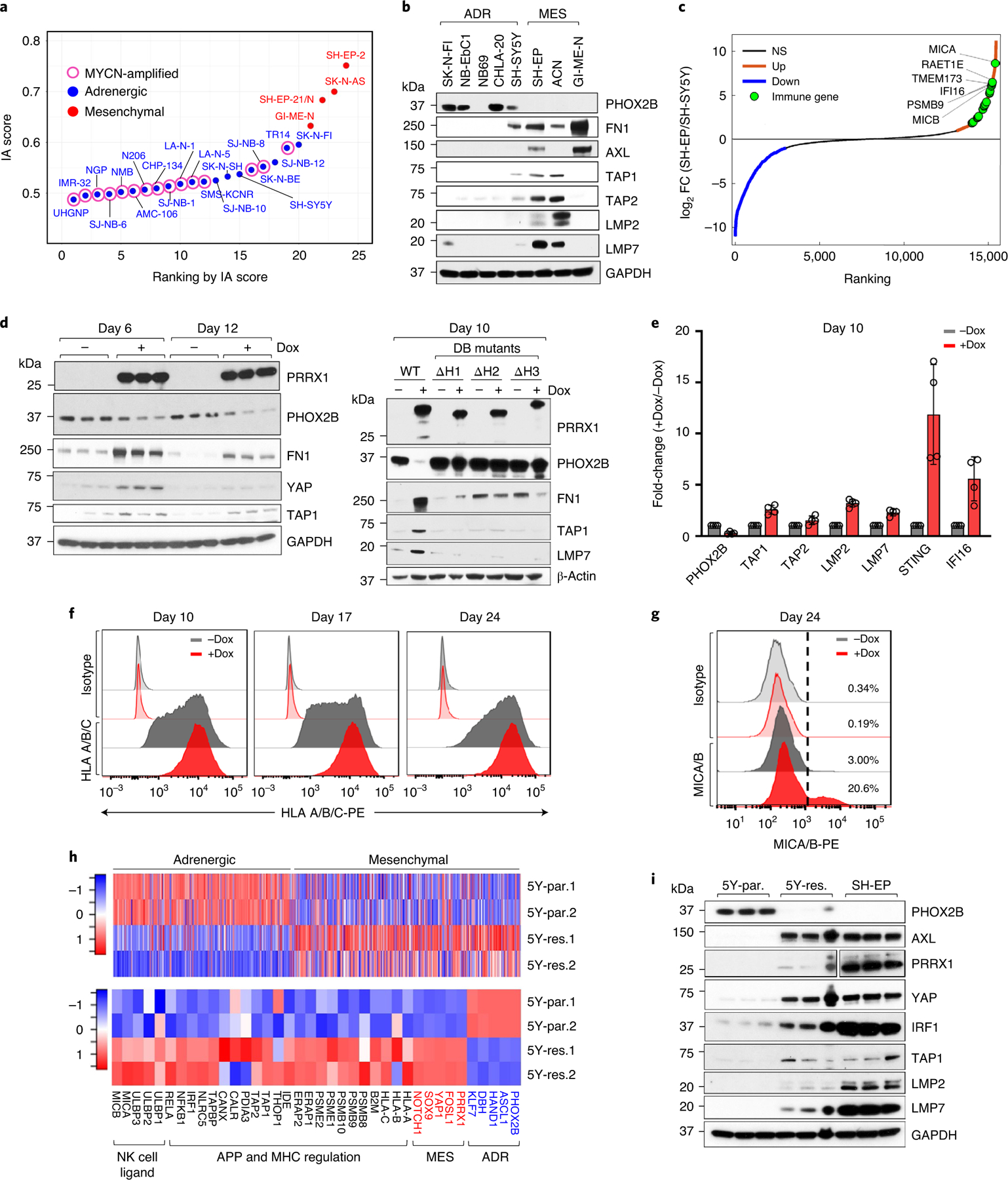
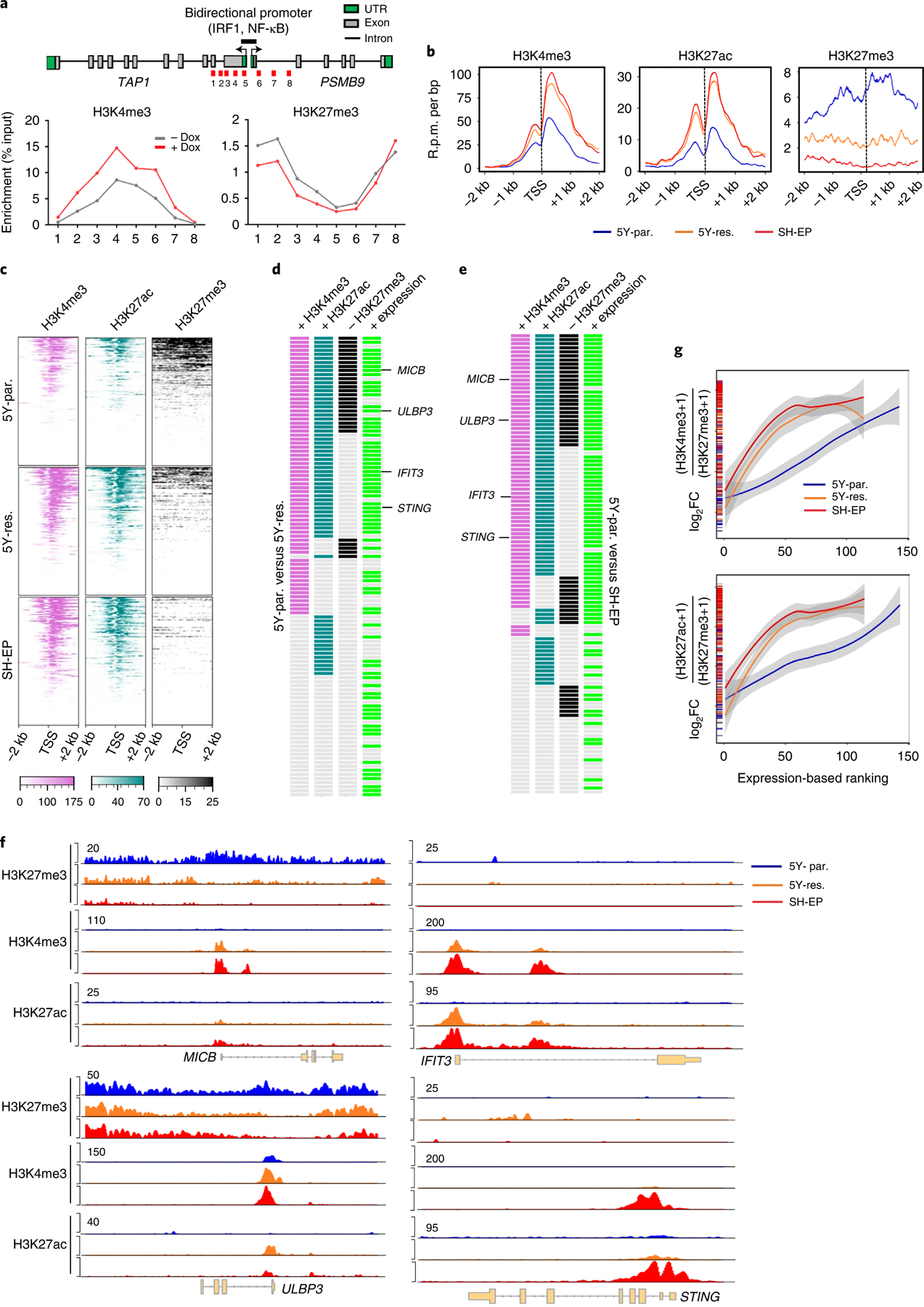
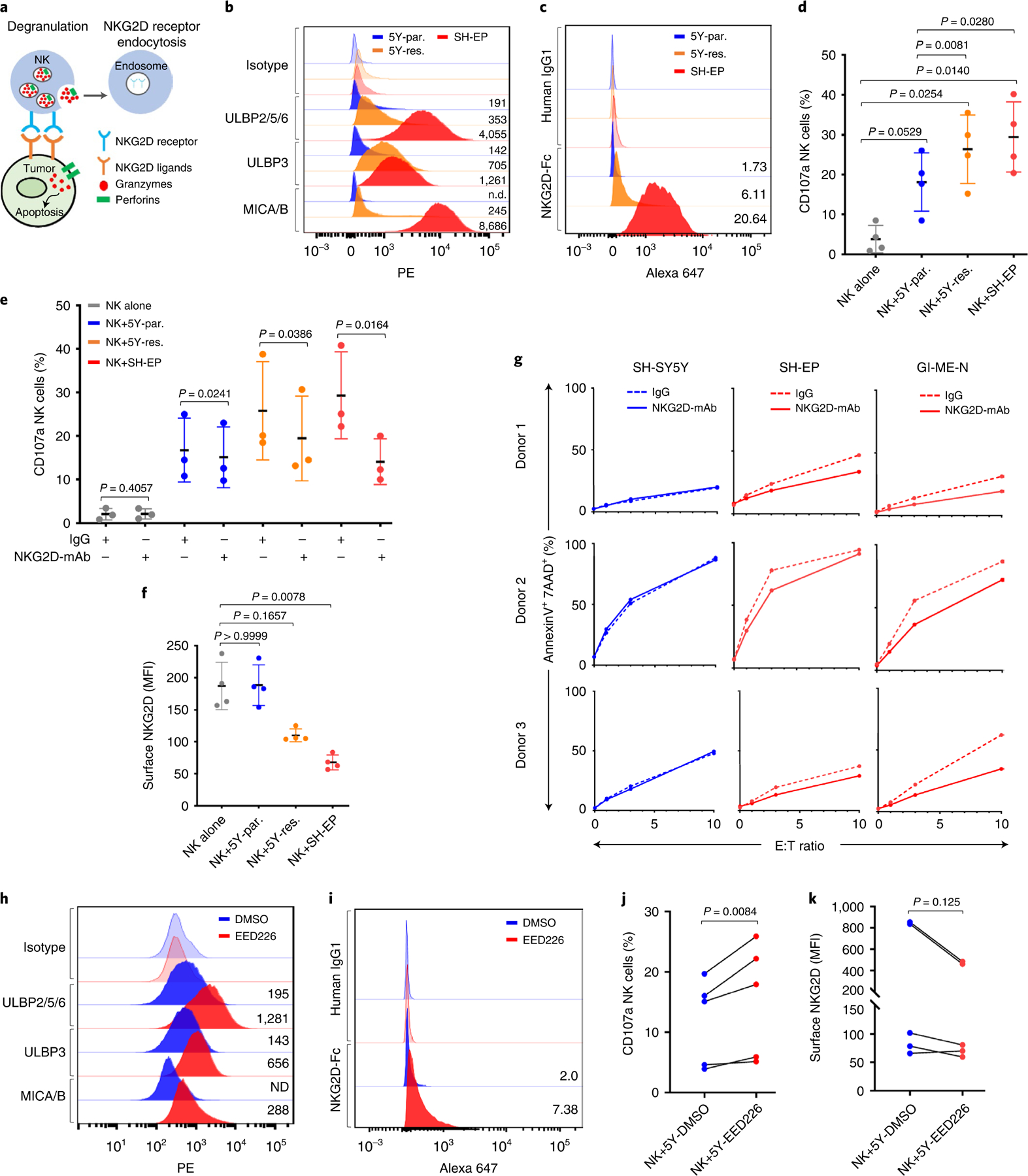
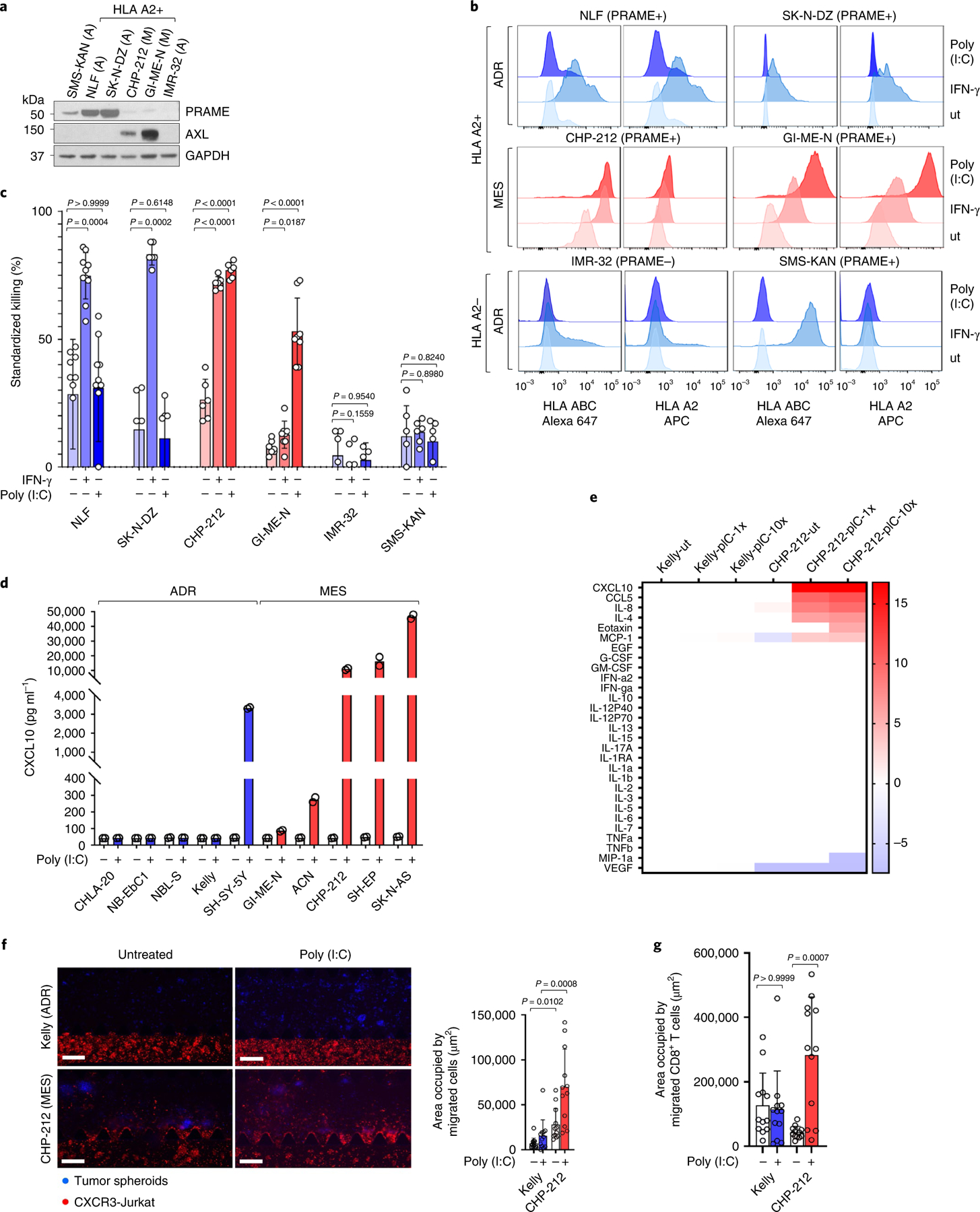
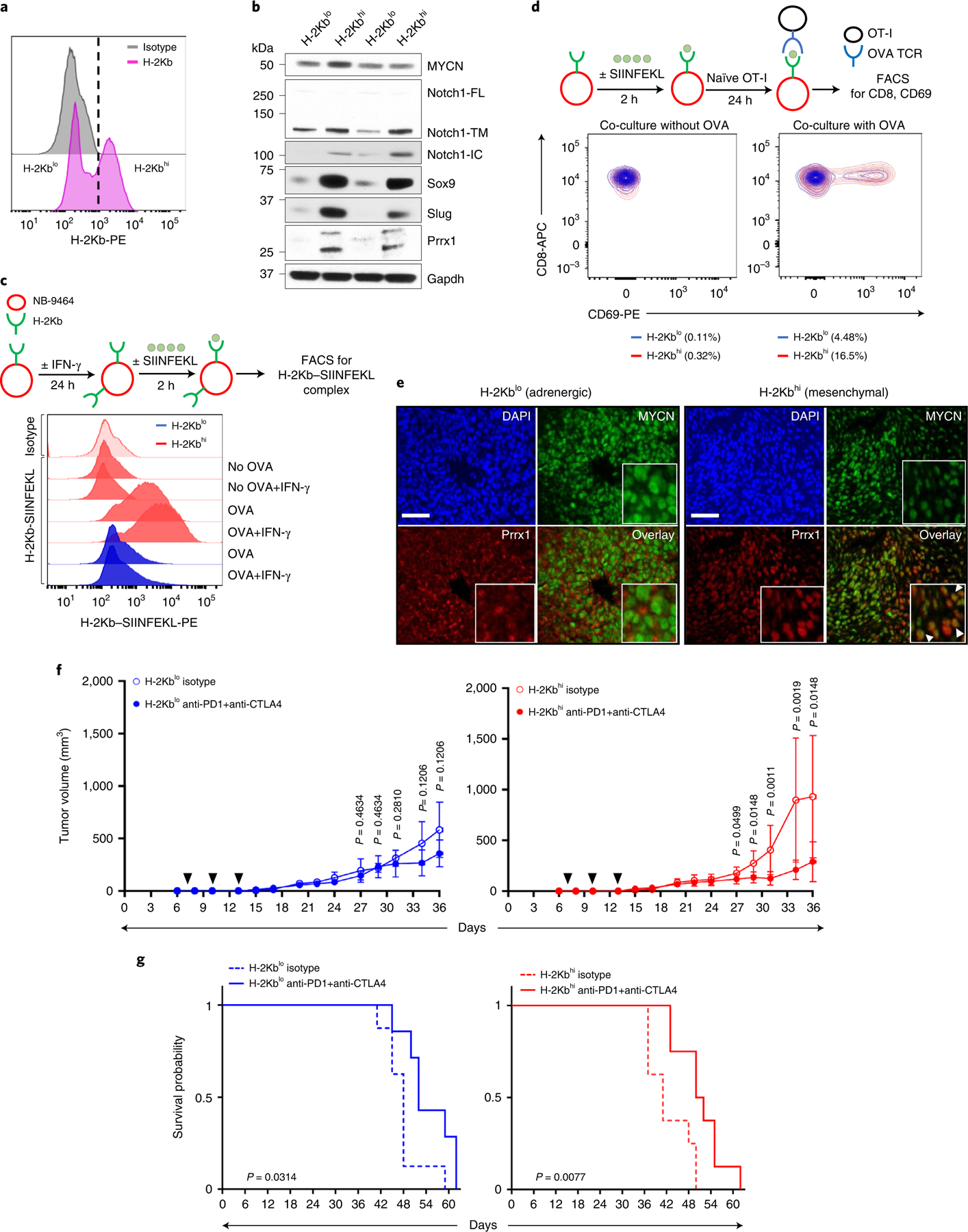
Apart from the anti-GD2 antibody, immunotherapy for neuroblastoma has had limited success due to immune evasion mechanisms, coupled with an incomplete understanding of predictors of response. Here, from bulk and single-cell transcriptomic analyses, we identify a subset of neuroblastomas enriched for transcripts associated with immune activation and inhibition and show that these are predominantly characterized by gene expression signatures of the mesenchymal lineage state. By contrast, tumors expressing adrenergic lineage signatures are less immunogenic. The inherent presence or induction of the mesenchymal state through transcriptional reprogramming or therapy resistance is accompanied by innate and adaptive immune gene activation through epigenetic remodeling. Mesenchymal lineage cells promote T cell infiltration by secreting inflammatory cytokines, are efficiently targeted by cytotoxic T and natural killer cells and respond to immune checkpoint blockade. Together, we demonstrate that distinct immunogenic phenotypes define the divergent lineage states of neuroblastoma and highlight the immunogenic potential of the mesenchymal lineage.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Barker E et al. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res 51, 144–149 (1991). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
