

Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation
- PMID: 36138229
- PMCID: PMC10068894
- DOI: 10.1038/s41588-022-01179-9
Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation
Abstract
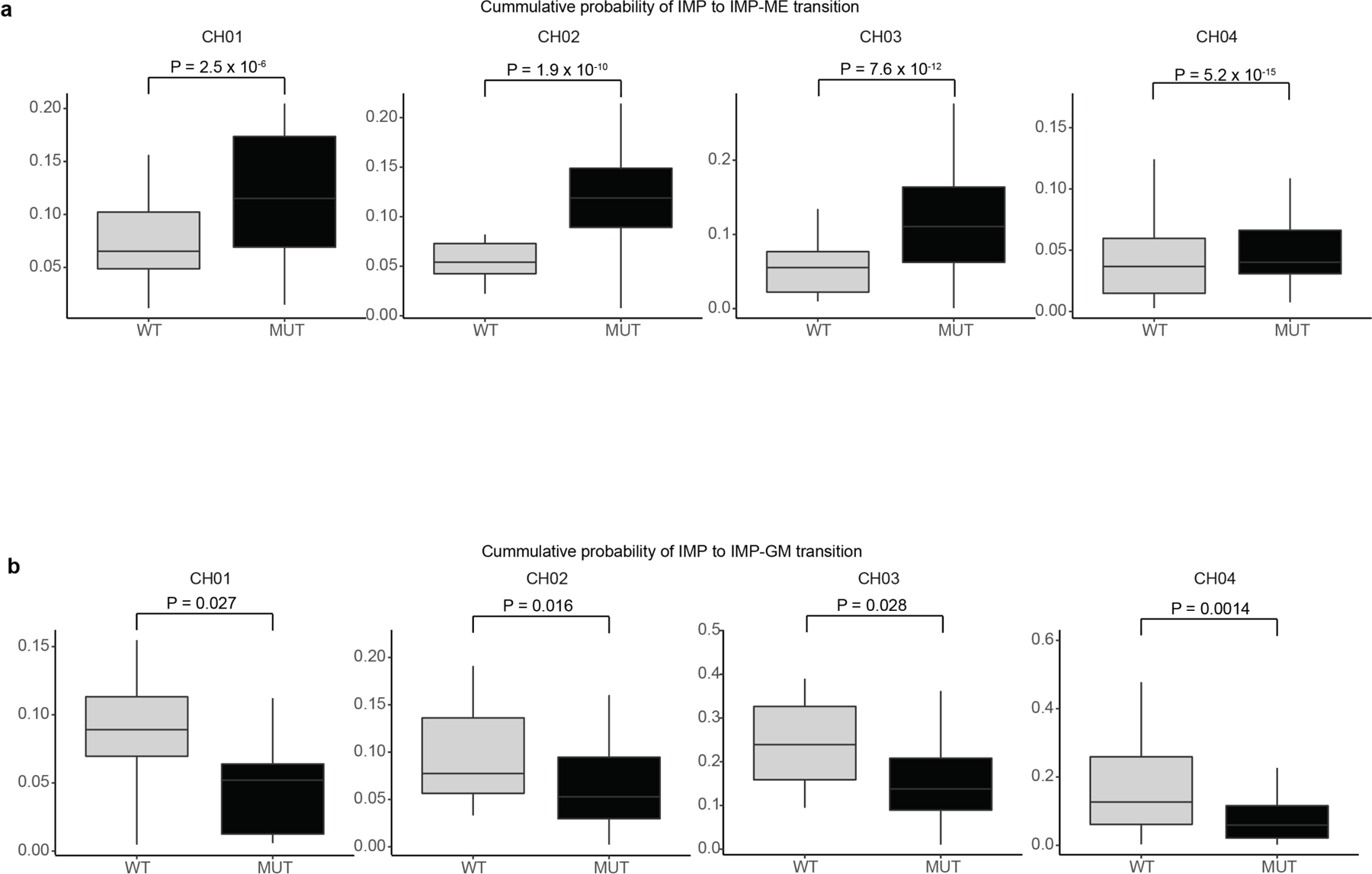
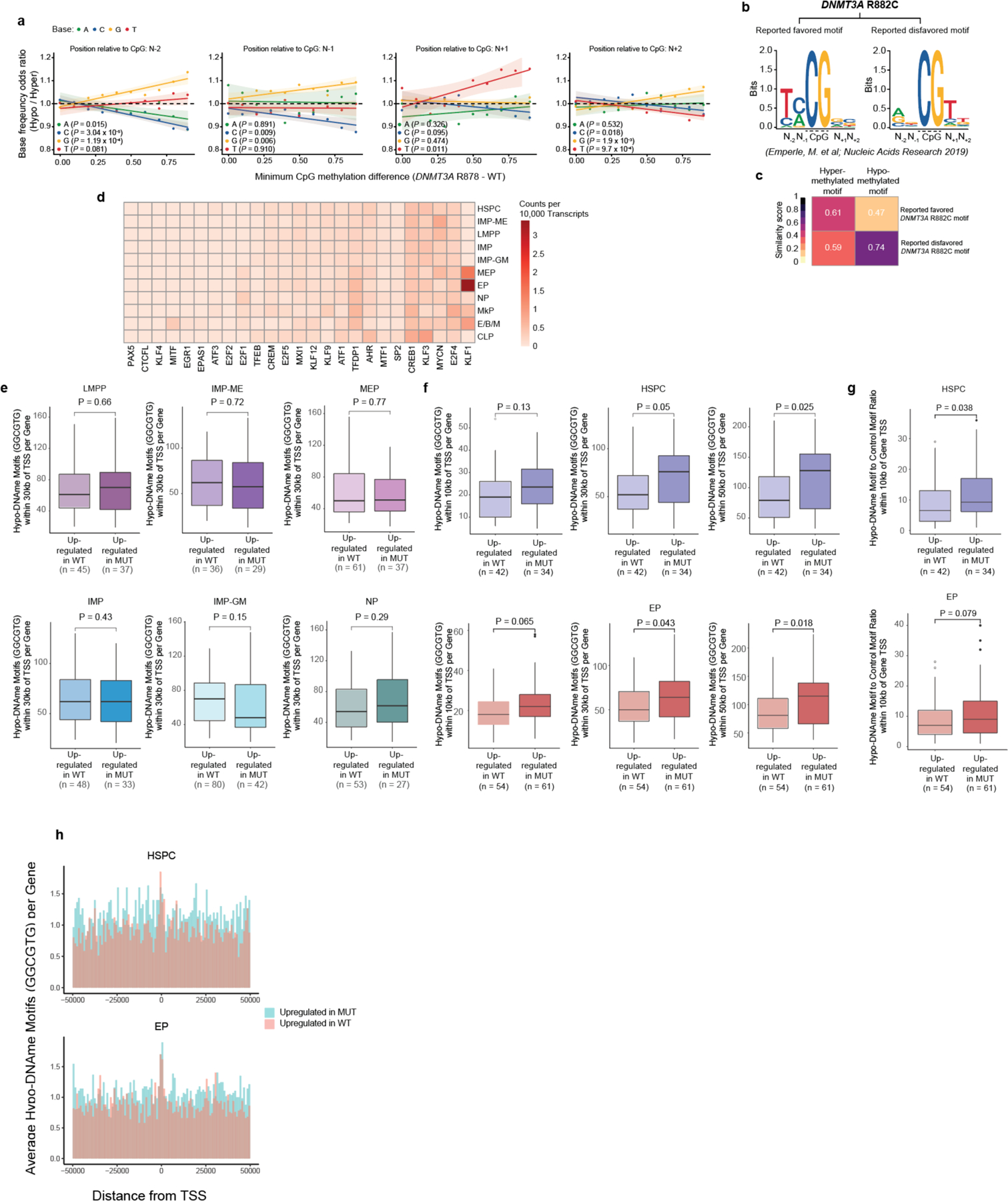
Somatic mutations in cancer genes have been detected in clonal expansions across healthy human tissue, including in clonal hematopoiesis. However, because mutated and wild-type cells are admixed, we have limited ability to link genotypes with phenotypes. To overcome this limitation, we leveraged multi-modality single-cell sequencing, capturing genotype, transcriptomes and methylomes in progenitors from individuals with DNMT3A R882 mutated clonal hematopoiesis. DNMT3A mutations result in myeloid over lymphoid bias, and an expansion of immature myeloid progenitors primed toward megakaryocytic-erythroid fate, with dysregulated expression of lineage and leukemia stem cell markers. Mutated DNMT3A leads to preferential hypomethylation of polycomb repressive complex 2 targets and a specific CpG flanking motif. Notably, the hypomethylation motif is enriched in binding motifs of key hematopoietic transcription factors, serving as a potential mechanistic link between DNMT3A mutations and aberrant transcriptional phenotypes. Thus, single-cell multi-omics paves the road to defining the downstream consequences of mutations that drive clonal mosaicism.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, Pfizer, and Janssen, and is on the Scientific Advisory Board of Envisagenics Inc and AIChemy; O.A.-W. has received prior research funding from H3B Biomedicine and LOXO Oncology unrelated to the current manuscript. I.G. serves on the advisory board of Bristol Myers Squibb, Takeda, Janssen, Sanofi and GlaxoSmithKline. D.A.L. has served as a consultant for Abbvie and Illumina, and is on the Scientific Advisory Board of Mission Bio and C2i Genomics; D.A.L. has received prior research funding from BMS and Illumina unrelated to the current manuscript.
Figures

















Comment in
-
Multi-omics on our multitudes.Nat Genet. 2022 Oct;54(10):1449-1450. doi: 10.1038/s41588-022-01175-z. Nat Genet. 2022. PMID: 36138230 No abstract available.
References
-
- Yokoyama A et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
