

SAIGE-GENE+ improves the efficiency and accuracy of set-based rare variant association tests
- PMID: 36138231
- PMCID: PMC9534766
- DOI: 10.1038/s41588-022-01178-w
SAIGE-GENE+ improves the efficiency and accuracy of set-based rare variant association tests
Erratum in
-
Publisher Correction: SAIGE-GENE+ improves the efficiency and accuracy of set-based rare variant association tests.Nat Genet. 2022 Nov;54(11):1755. doi: 10.1038/s41588-022-01220-x. Nat Genet. 2022. PMID: 36257984 Free PMC article. No abstract available.
Abstract
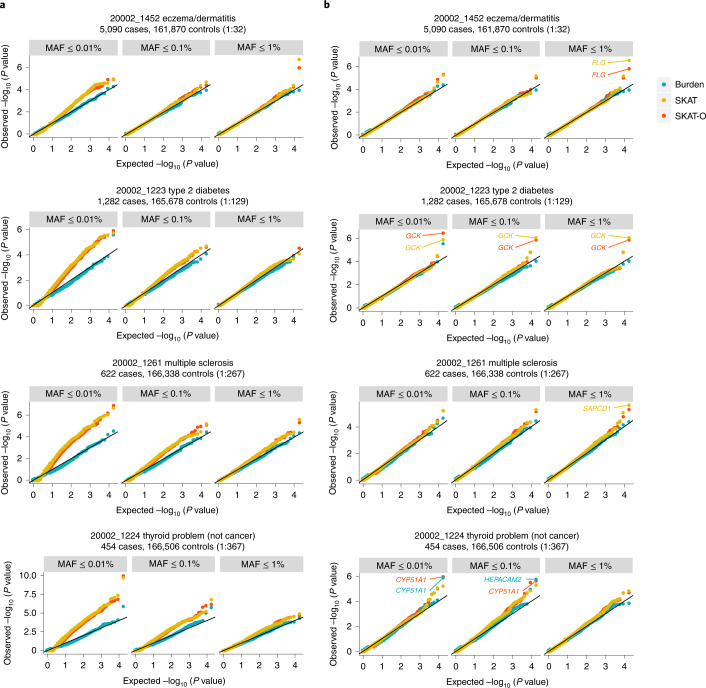
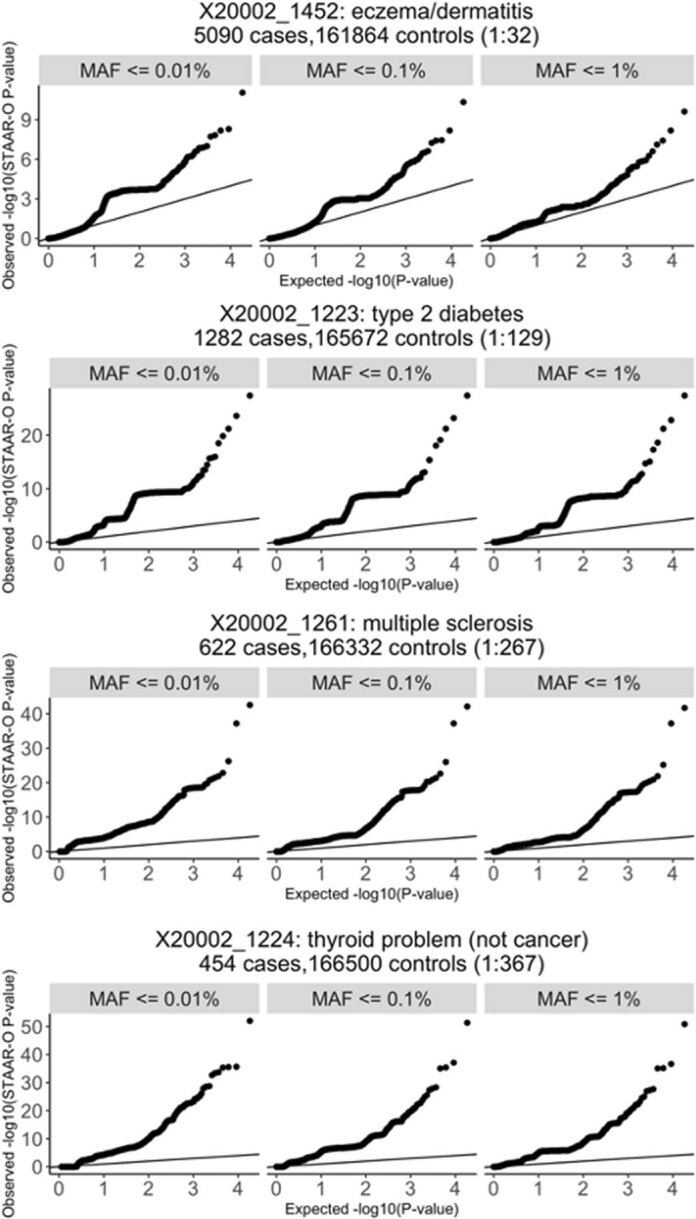
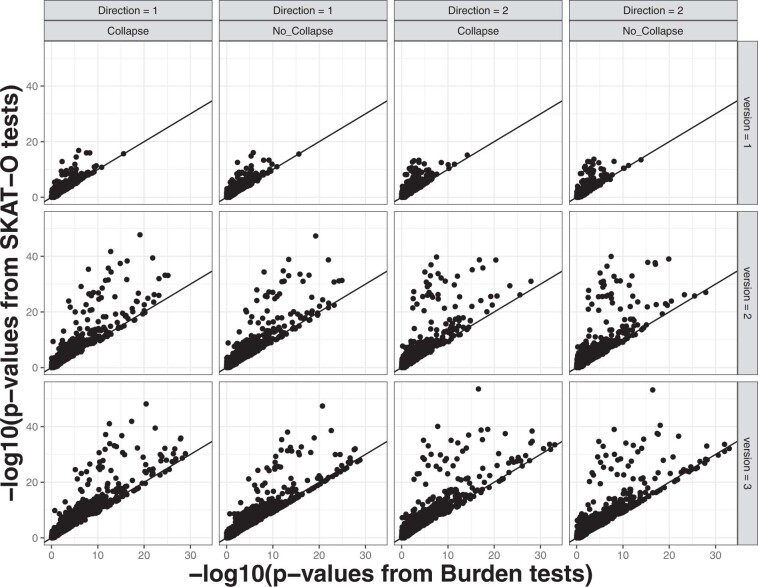
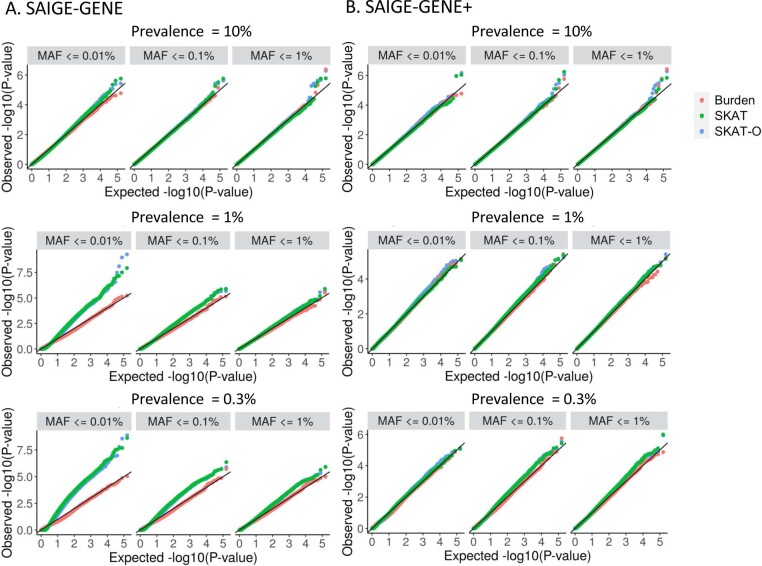
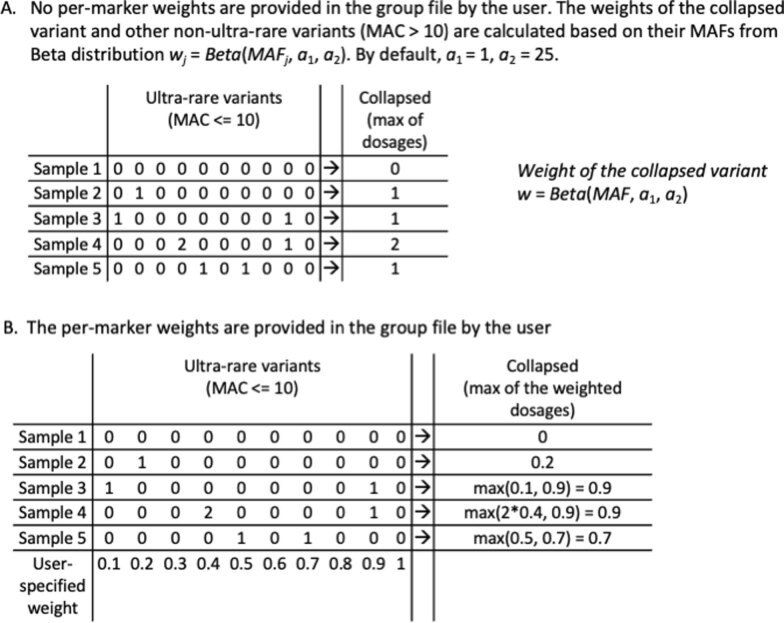
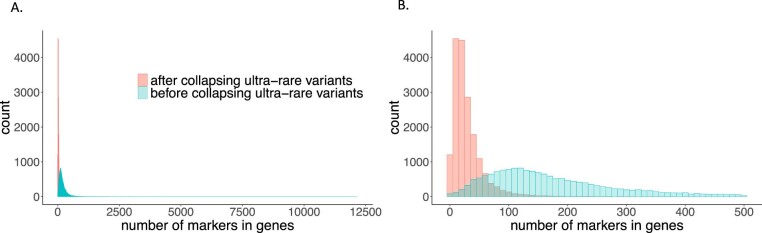
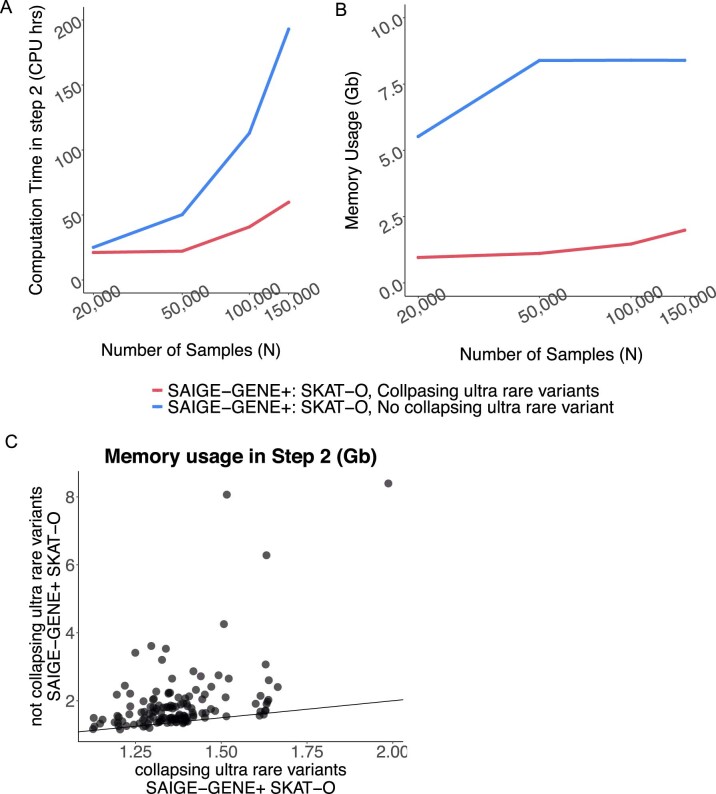
Several biobanks, including UK Biobank (UKBB), are generating large-scale sequencing data. An existing method, SAIGE-GENE, performs well when testing variants with minor allele frequency (MAF) ≤ 1%, but inflation is observed in variance component set-based tests when restricting to variants with MAF ≤ 0.1% or 0.01%. Here, we propose SAIGE-GENE+ with greatly improved type I error control and computational efficiency to facilitate rare variant tests in large-scale data. We further show that incorporating multiple MAF cutoffs and functional annotations can improve power and thus uncover new gene-phenotype associations. In the analysis of UKBB whole exome sequencing data for 30 quantitative and 141 binary traits, SAIGE-GENE+ identified 551 gene-phenotype associations.
© 2022. The Author(s).
Conflict of interest statement
B.M.N. is a member of Deep Genomics Scientific Advisory Board, has received travel expenses from Illumina, and also serves as a consultant for Avanir and Trigeminal solutions. K.J.K. is a consultant for Vor Biopharma. The remaining authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
