

The Clinical Management of Pompe Disease: A Pediatric Perspective
- PMID: 36138713
- PMCID: PMC9497581
- DOI: 10.3390/children9091404
The Clinical Management of Pompe Disease: A Pediatric Perspective
Abstract
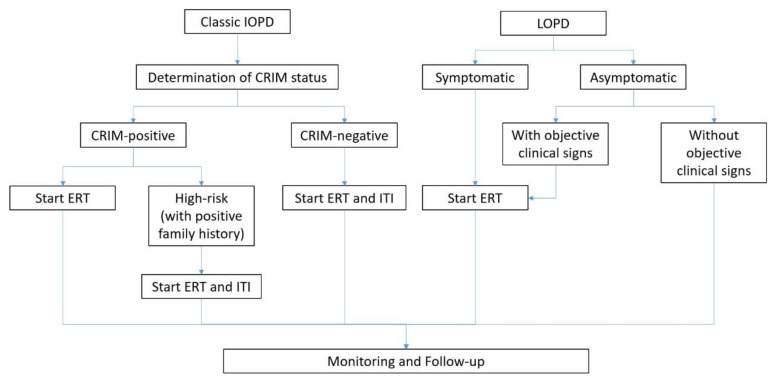
Pompe disease (PD) is an inherited metabolic disorder caused by a deficiency of acid α-glucosidase (GAA), leading to lysosomal accumulation of glycogen, mainly in skeletal and cardiac muscles as well as the nervous system. Patients with PD develop cellular dysfunction and muscle damage. PD can be classified into two classic forms, namely infantile-onset PD (IOPD) and late-onset PD (LOPD). Delayed treatment, particularly in IOPD, would result in significant organ damage and early death. Nonetheless, early diagnosis and timely treatment are often hampered by the rarity of PD and its wide variety of, but overlapping, symptoms. This article reviews the common clinical presentations of PD and outlines the essentials of PD management. In particular, the implications of newborn screening (NBS) and clinical performance of enzyme replacement therapy (ERT) are highlighted.
Keywords: Pompe disease; alglucosidase alpha; clinical management; enzyme replacement therapy; newborn screening.
Conflict of interest statement
The authors declare no conflict of interest.
References
-
- Kronn D.F., Day-Salvatore D., Hwu W.-L., Jones S.A., Nakamura K., Okuyama T., Swoboda K.J., Kishnani P.S., on behalf of the Pompe Disease Newborn Screening Working Group Management of Confirmed Newborn-Screened Patients with Pompe Disease Across the Disease Spectrum. Pediatrics. 2017;140((Suppl. 1)):S24–S45. doi: 10.1542/peds.2016-0280E. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
