

Integrin-Linked Kinase Expression in Human Valve Endothelial Cells Plays a Protective Role in Calcific Aortic Valve Disease
- PMID: 36139812
- PMCID: PMC9495882
- DOI: 10.3390/antiox11091736
Integrin-Linked Kinase Expression in Human Valve Endothelial Cells Plays a Protective Role in Calcific Aortic Valve Disease
Abstract
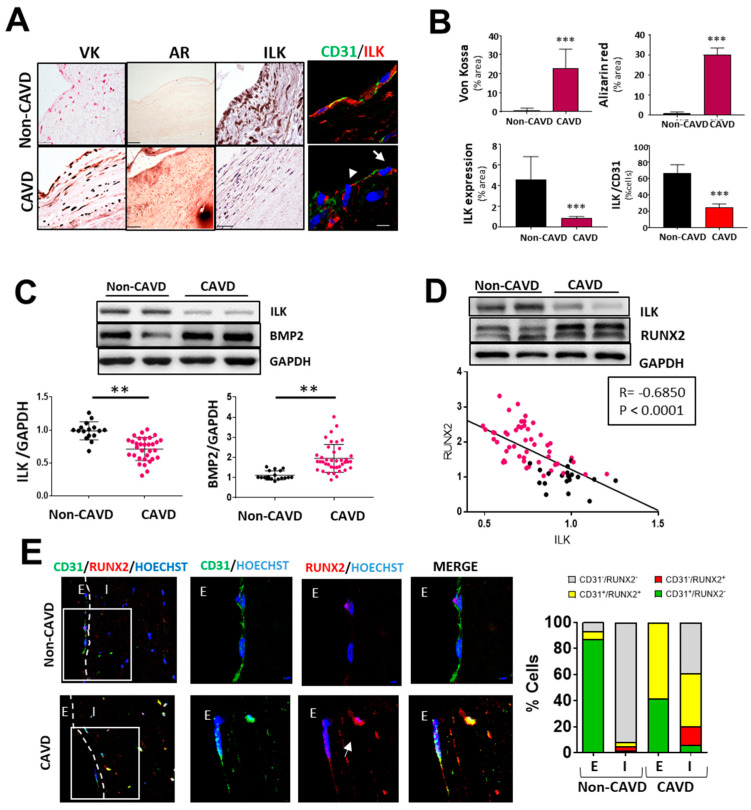
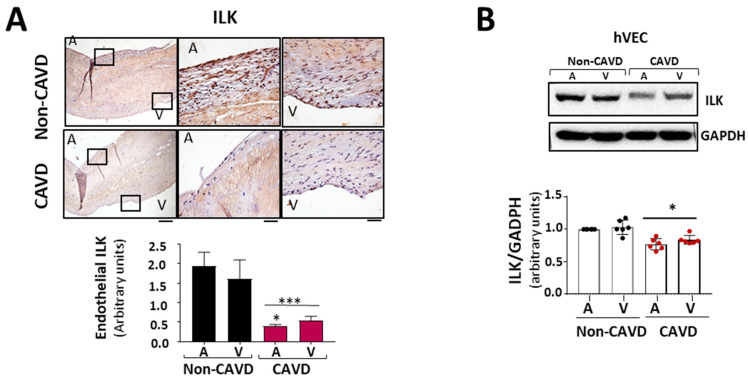
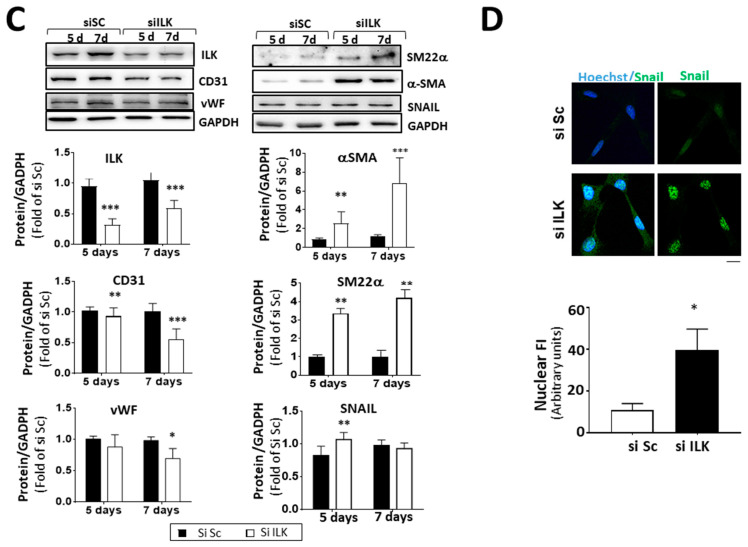
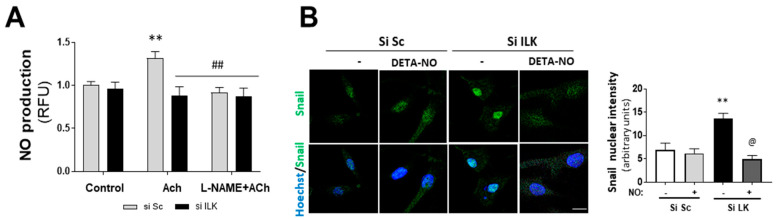
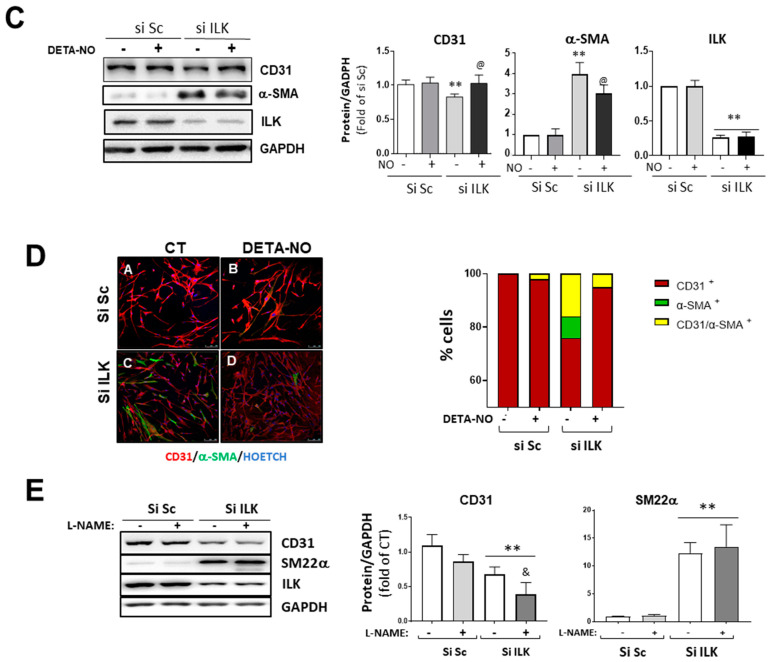
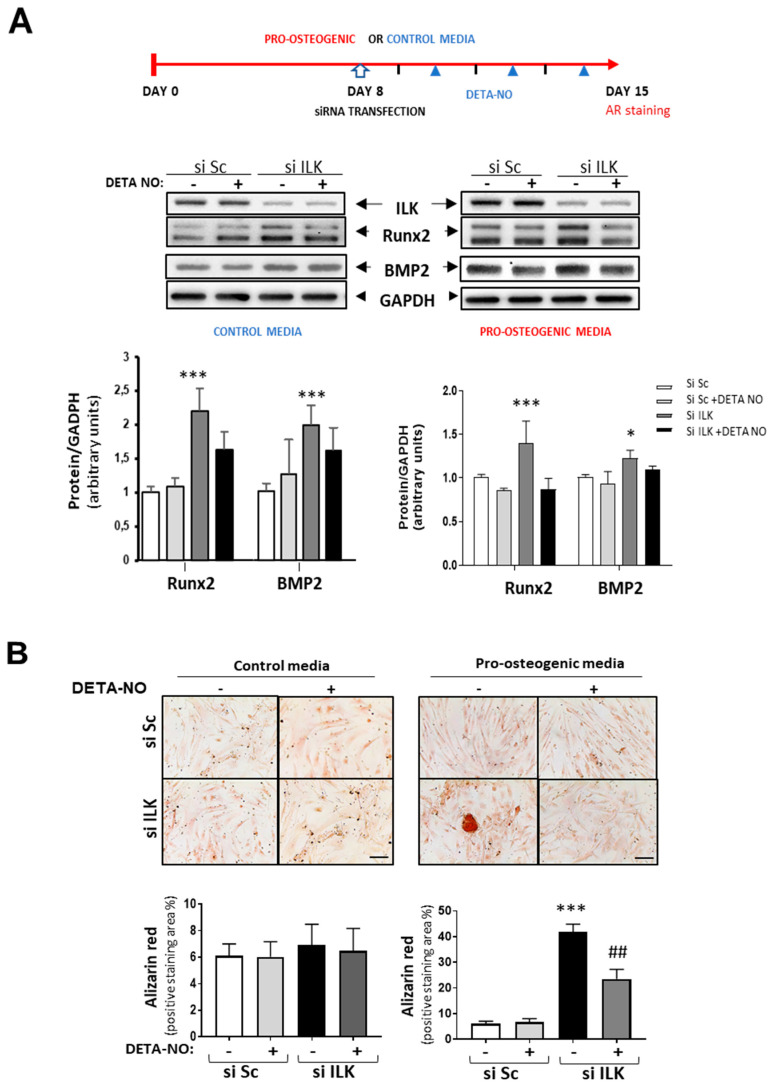
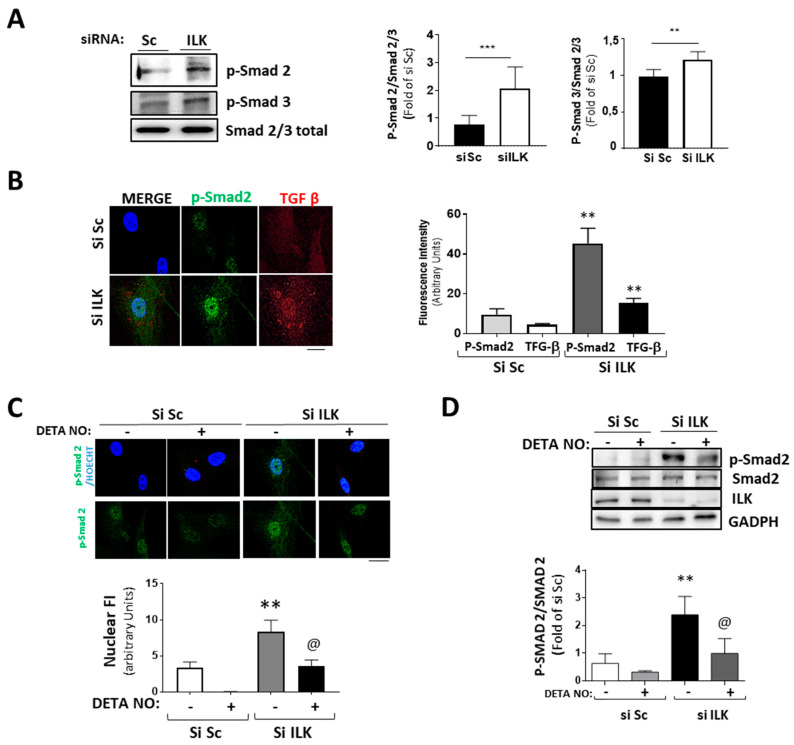
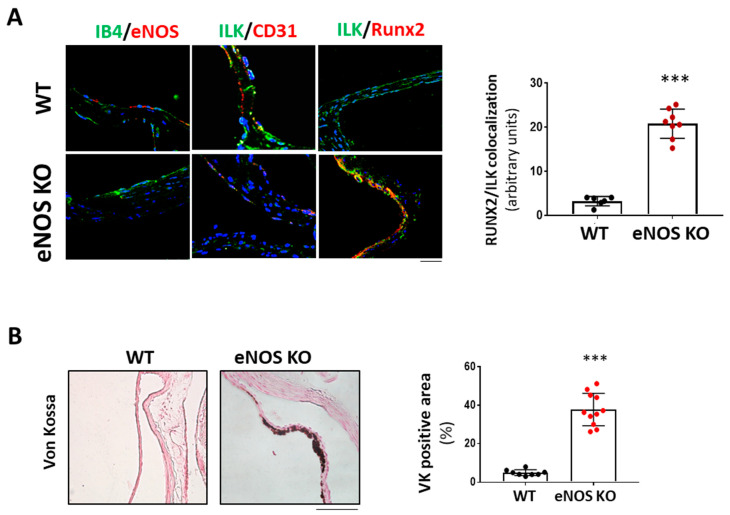
Calcific aortic valve disease (CAVD) is highly prevalent during aging. CAVD initiates with endothelial dysfunction, leading to lipid accumulation, inflammation, and osteogenic transformation. Integrin-linked kinase (ILK) participates in the progression of cardiovascular diseases, such as endothelial dysfunction and atherosclerosis. However, ILK role in CAVD is unknown. First, we determined that ILK expression is downregulated in aortic valves from patients with CAVD compared to non-CAVD, especially at the valve endothelium, and negatively correlated with calcification markers. Silencing ILK expression in human valve endothelial cells (siILK-hVECs) induced endothelial-to-mesenchymal transition (EndMT) and promoted a switch to an osteoblastic phenotype; SiILK-hVECs expressed increased RUNX2 and developed calcified nodules. siILK-hVECs exhibited decreased NO production and increased nitrosative stress, suggesting valvular endothelial dysfunction. NO treatment of siILK-hVECs prevented VEC transdifferentiation, while treatment with an eNOS inhibitor mimicked ILK-silencing induction of EndMT. Accordingly, NO treatment inhibited VEC calcification. Mechanistically, siILK-hVECs showed increased Smad2 phosphorylation, suggesting a TGF-β-dependent mechanism, and NO treatment decreased Smad2 activation and RUNX2. Experiments performed in eNOS KO mice confirmed the involvement of the ILK-eNOS signaling pathway in valve calcification, since aortic valves from these animals showed decreased ILK expression, increased RUNX2, and calcification. Our study demonstrated that ILK endothelial expression participates in human CAVD development by preventing endothelial osteogenic transformation.
Keywords: EndMT; ILK; aging; calcific valve disease; endothelial dysfunction; nitric oxide.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Yadgir S., Johnson C.O., Aboyans V., Adebayo O.M., Adedoyin R.A., Afarideh M., Alahdab F., Alashi A., Alipour V., Arabloo J., et al. Global, Regional, and National Burden of Calcific Aortic Valve and Degenerative Mitral Valve Diseases, 1990–2017. Circulation. 2020;141:1670–1680. doi: 10.1161/CIRCULATIONAHA.119.043391. - DOI - PubMed
-
- Lindman B.R., Sukul D., Dweck M.R., Madhavan M.V., Arsenault B.J., Coylewright M., Merryman W.D., Newby D.E., Lewis J., Harrell F.E., et al. Evaluating Medical Therapy for Calcific Aortic Stenosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021;78:2354–2376. doi: 10.1016/j.jacc.2021.09.1367. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
