

Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2
- PMID: 36142405
- PMCID: PMC9499601
- DOI: 10.3390/ijms231810491
Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2
Abstract
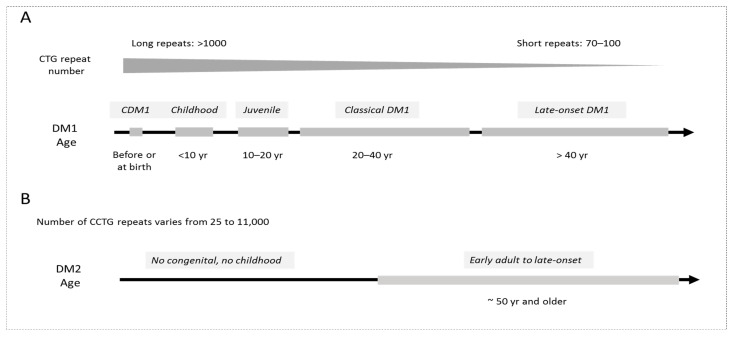
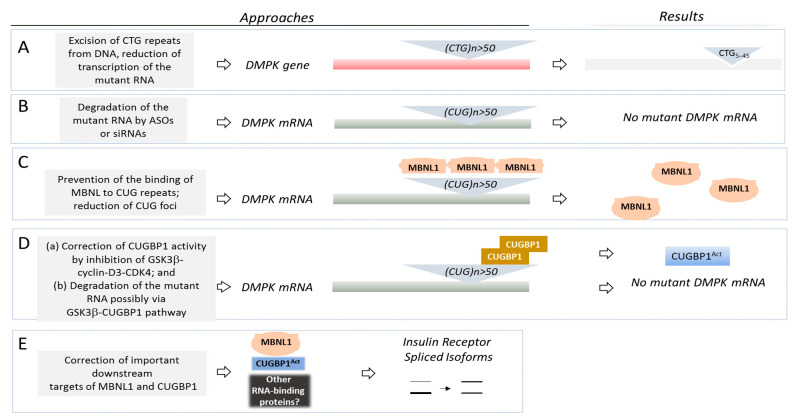
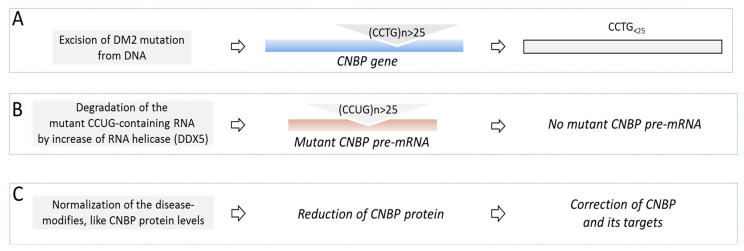
Myotonic Dystrophies type 1 (DM1) and type 2 (DM2) are complex multisystem diseases without disease-based therapies. These disorders are caused by the expansions of unstable CTG (DM1) and CCTG (DM2) repeats outside of the coding regions of the disease genes: DMPK in DM1 and CNBP in DM2. Multiple clinical and molecular studies provided a consensus for DM1 pathogenesis, showing that the molecular pathophysiology of DM1 is associated with the toxicity of RNA CUG repeats, which cause multiple disturbances in RNA metabolism in patients' cells. As a result, splicing, translation, RNA stability and transcription of multiple genes are misregulated in DM1 cells. While mutant CCUG repeats are the main cause of DM2, additional factors might play a role in DM2 pathogenesis. This review describes current progress in the translation of mechanistic knowledge in DM1 and DM2 to clinical trials, with a focus on the development of disease-specific therapies for patients with adult forms of DM1 and congenital DM1 (CDM1).
Keywords: clinical trials; congenital myotonic dystrophy; myotonic dystrophy; myotonic dystrophy type 2.
Conflict of interest statement
The author’s research related to tideglusib studies was partially supported by funding from AMO Pharma. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
Similar articles
-
(CCUG)n RNA toxicity in a Drosophila model of myotonic dystrophy type 2 (DM2) activates apoptosis.Dis Model Mech. 2017 Aug 1;10(8):993-1003. doi: 10.1242/dmm.026179. Epub 2017 Jun 16. Dis Model Mech. 2017. PMID: 28623239 Free PMC article.
-
Molecular mechanisms of muscle atrophy in myotonic dystrophies.Int J Biochem Cell Biol. 2013 Oct;45(10):2280-7. doi: 10.1016/j.biocel.2013.06.010. Epub 2013 Jun 21. Int J Biochem Cell Biol. 2013. PMID: 23796888 Free PMC article. Review.
-
Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies.Acta Myol. 2013 Dec;32(3):154-65. Acta Myol. 2013. PMID: 24803843 Free PMC article. Review.
-
CCUG repeats reduce the rate of global protein synthesis in myotonic dystrophy type 2.Rev Neurosci. 2010;21(1):19-28. doi: 10.1515/revneuro.2010.21.1.19. Rev Neurosci. 2010. PMID: 20458885 Review.
-
Expanded CCUG repeat RNA expression in Drosophila heart and muscle trigger Myotonic Dystrophy type 1-like phenotypes and activate autophagocytosis genes.Sci Rep. 2017 Jun 6;7(1):2843. doi: 10.1038/s41598-017-02829-3. Sci Rep. 2017. PMID: 28588248 Free PMC article.
Cited by
-
Pluripotent Stem Cells in Disease Modeling and Drug Discovery for Myotonic Dystrophy Type 1.Cells. 2023 Feb 10;12(4):571. doi: 10.3390/cells12040571. Cells. 2023. PMID: 36831237 Free PMC article. Review.
-
Resistance Exercise Training Rescues Mitochondrial Dysfunction in Skeletal Muscle of Patients with Myotonic Dystrophy Type 1.J Neuromuscul Dis. 2023;10(6):1111-1126. doi: 10.3233/JND-230099. J Neuromuscul Dis. 2023. PMID: 37638448 Free PMC article.
-
Huntington disease-like 2: insight into neurodegeneration from an African disease.Nat Rev Neurol. 2024 Jan;20(1):36-49. doi: 10.1038/s41582-023-00906-y. Epub 2023 Dec 19. Nat Rev Neurol. 2024. PMID: 38114648 Review.
-
Expert Insights from a Delphi-driven Neurologists' Panel: Real-world Mexiletine use in Patients with Myotonic Disorders in Italy.J Neuromuscul Dis. 2024;11(2):411-423. doi: 10.3233/JND-230115. J Neuromuscul Dis. 2024. PMID: 38306059 Free PMC article.
-
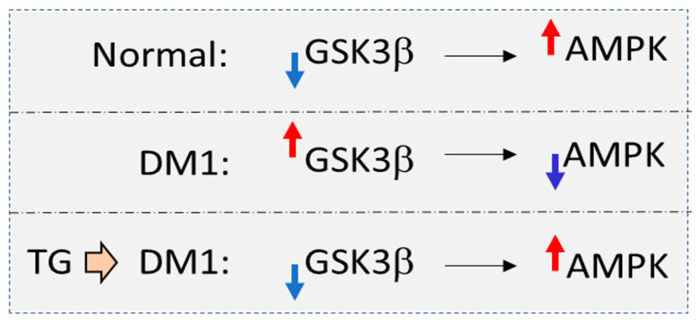
Therapeutic Targeting of the GSK3β-CUGBP1 Pathway in Myotonic Dystrophy.Int J Mol Sci. 2023 Jun 26;24(13):10650. doi: 10.3390/ijms241310650. Int J Mol Sci. 2023. PMID: 37445828 Free PMC article.
References
-
- Liquori C.L., Ricker K., Moseley M.L., Jacobsen J.F., Kress W., Naylor S.L., Day J.W., Ranum L.P. Myotonic dystrophy 2 is caused by CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. - PubMed
-
- Harper P.S. Myotonic Dystrophy. WB Saunders; London, UK: 2001.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
