

Glial Cell-Mediated Neuroinflammation in Alzheimer's Disease
- PMID: 36142483
- PMCID: PMC9502483
- DOI: 10.3390/ijms231810572
Glial Cell-Mediated Neuroinflammation in Alzheimer's Disease
Abstract
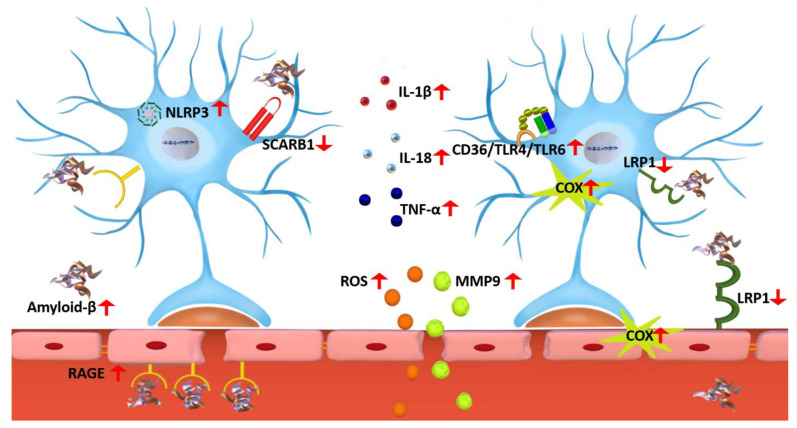
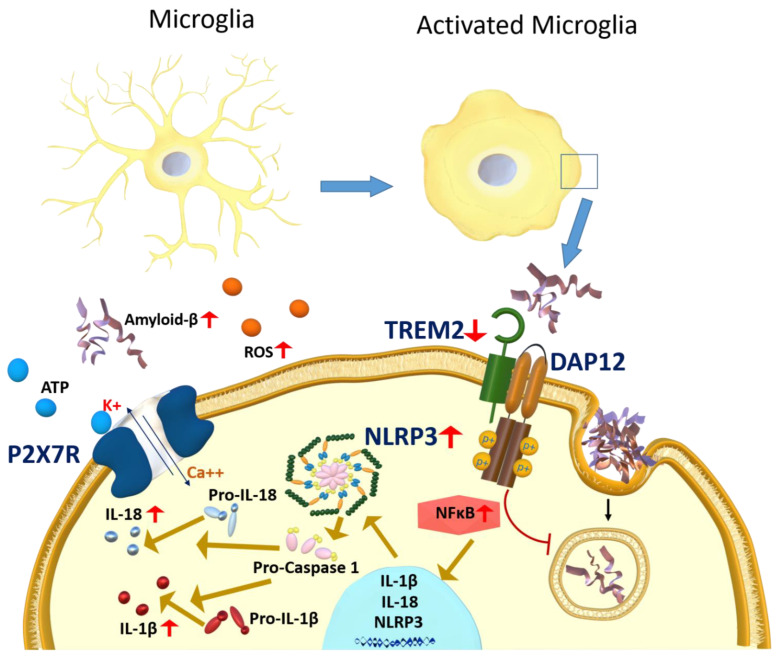
Alzheimer's disease (AD) is a progressive neurodegenerative disorder; it is the most common cause of dementia and has no treatment. It is characterized by two pathological hallmarks, the extracellular deposits of amyloid beta (Aβ) and the intraneuronal deposits of Neurofibrillary tangles (NFTs). Yet, those two hallmarks do not explain the full pathology seen with AD, suggesting the involvement of other mechanisms. Neuroinflammation could offer another explanation for the progression of the disease. This review provides an overview of recent advances on the role of the immune cells' microglia and astrocytes in neuroinflammation. In AD, microglia and astrocytes become reactive by several mechanisms leading to the release of proinflammatory cytokines that cause further neuronal damage. We then provide updates on neuroinflammation diagnostic markers and investigational therapeutics currently in clinical trials to target neuroinflammation.
Keywords: Alzheimer’s disease; astrocytes; clinical trials; diagnostic markers; microglia; neuroinflammation; therapeutics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
