

AST-487 Inhibits RET Kinase Driven TERT Expression in Bladder Cancer
- PMID: 36142729
- PMCID: PMC9501578
- DOI: 10.3390/ijms231810819
AST-487 Inhibits RET Kinase Driven TERT Expression in Bladder Cancer
Abstract
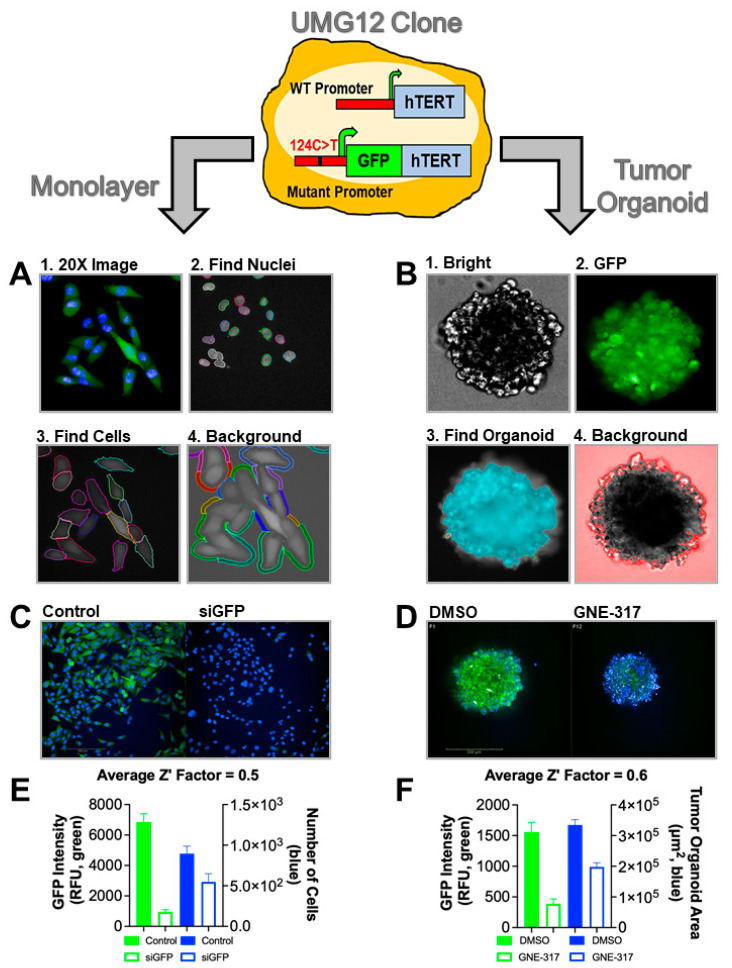
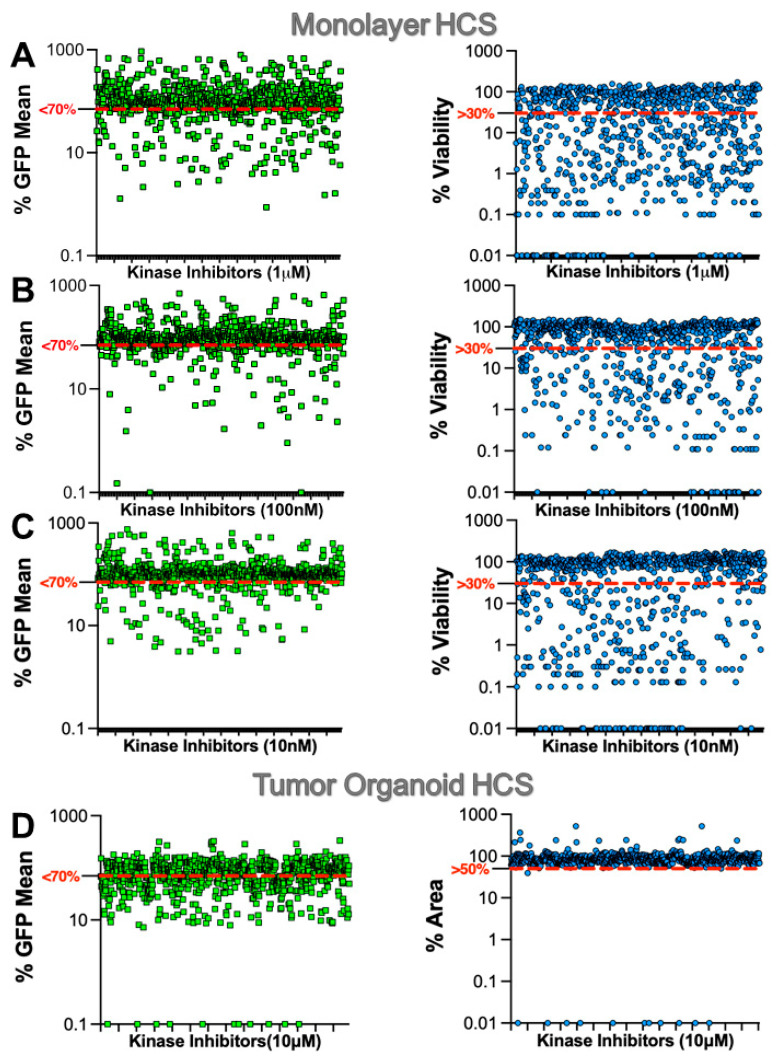
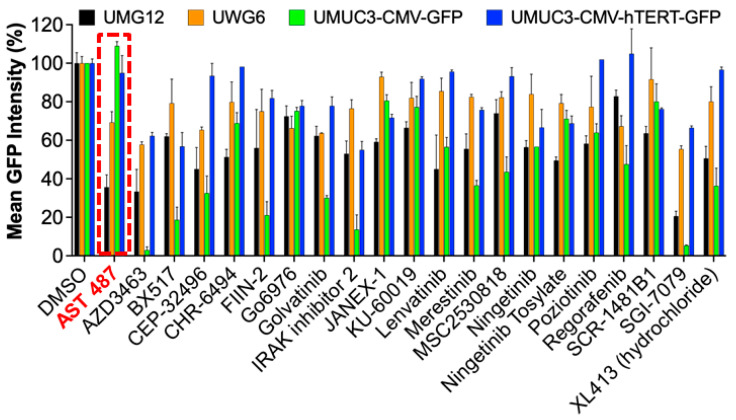
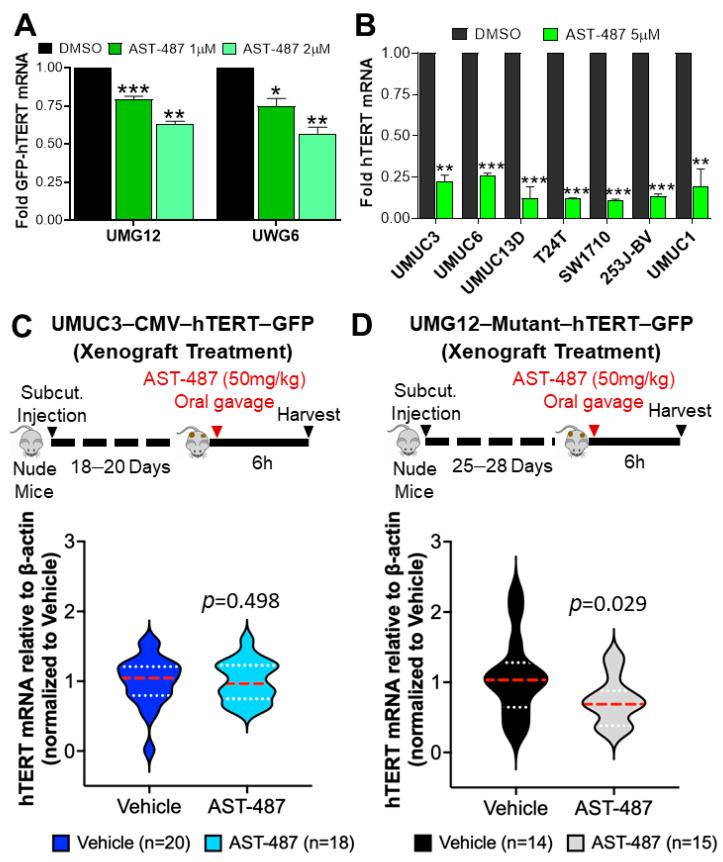
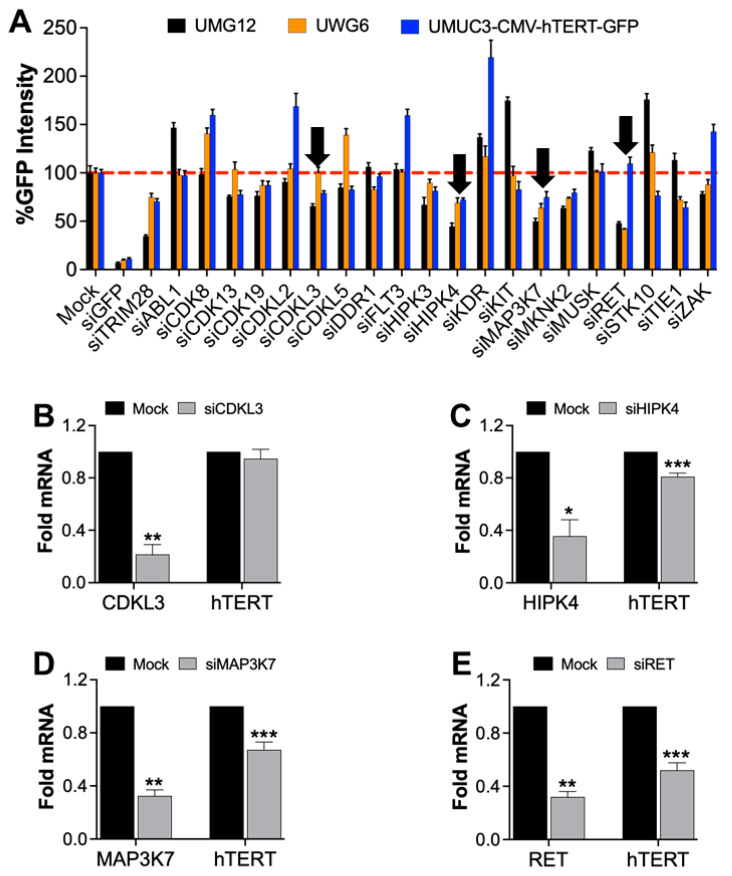
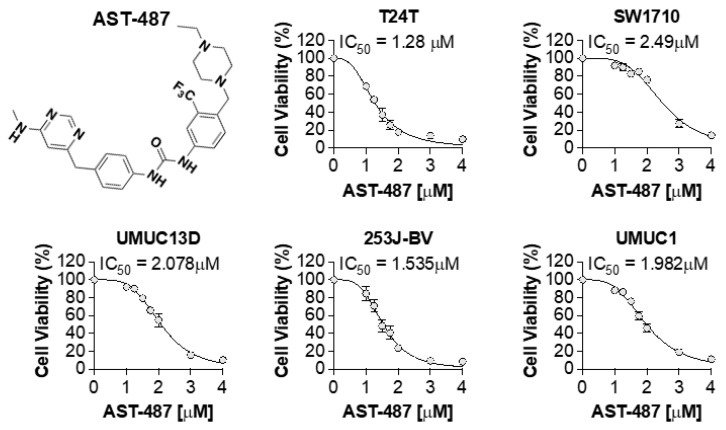
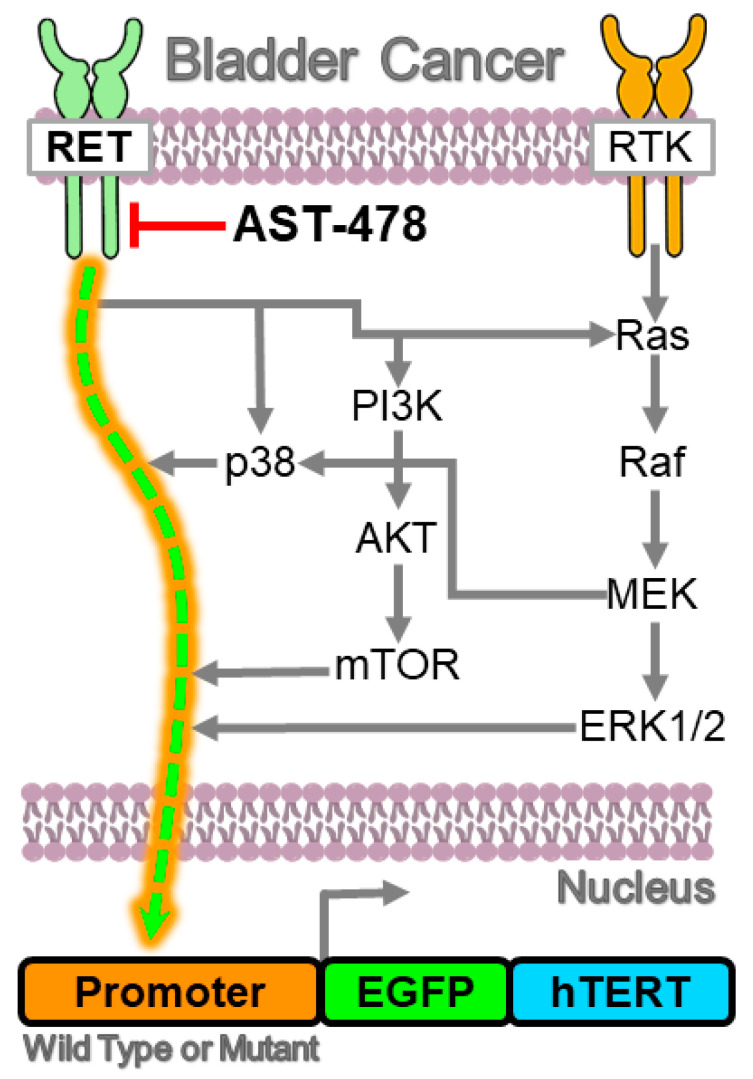
Mutations in the promoter of the human Telomerase Reverse Transcriptase (hTERT) gene are common and associated with its elevated expression in bladder cancer, melanoma, and glioblastoma. Though these mutations and TERT overexpression are associated with aggressive disease and poor outcome, an incomplete understanding of mutant TERT regulation limits treatment options directed at this gene. Herein, we unravel a signaling pathway that leads to upregulated hTERT expression resulting from the -124 bp promoter mutation, the most frequent variant across human cancer. We employed engineered bladder cancer cells that harbor a GFP insertion at the TSS region on -124 hTERT promoter for high-content screening drug discovery using a focused library of ~800 kinase inhibitors. Studies using in vitro and in vivo models prioritized AST-487, an inhibitor of the wild-type, and mutant RET (rearranged during transfection) proto-oncogene as a novel drug inhibitor of both wild-type and mutant promoter-driven hTERT expression. We also identified the RET kinase pathway, targeted by AST-487, as a novel regulator of mutant hTERT promoter-driven transcription in bladder cancer cells. Collectively, our work provides new potential precision medicine approaches for cancer patients with upregulated hTERT expression, perhaps, especially those harboring mutations in both the RET gene and the hTERT promoter, such as in thyroid cancer.
Keywords: RET; TERT; telomerase.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
TRIM28 is a transcriptional activator of the mutant TERT promoter in human bladder cancer.Proc Natl Acad Sci U S A. 2021 Sep 21;118(38):e2102423118. doi: 10.1073/pnas.2102423118. Proc Natl Acad Sci U S A. 2021. PMID: 34518220 Free PMC article.
-
Synthetic Bax-Anti Bcl2 combination module actuated by super artificial hTERT promoter selectively inhibits malignant phenotypes of bladder cancer.J Exp Clin Cancer Res. 2016 Jan 8;35:3. doi: 10.1186/s13046-015-0279-6. J Exp Clin Cancer Res. 2016. PMID: 26743236 Free PMC article.
-
hTERT transcription is repressed by Cbfa1 in human mesenchymal stem cell populations.J Bone Miner Res. 2007 Jun;22(6):897-906. doi: 10.1359/jbmr.070308. J Bone Miner Res. 2007. PMID: 17352650
-
hTERT, hTR and TERT promoter mutations as markers for urological cancers detection: A systematic review.Urol Oncol. 2021 Aug;39(8):498.e21-498.e33. doi: 10.1016/j.urolonc.2021.01.022. Epub 2021 Mar 3. Urol Oncol. 2021. PMID: 33676848
-
Telomerase activity, TERT expression, hTERT promoter alterations, and alternative lengthening of the telomeres (ALT) in meningiomas - a systematic review.Neurosurg Rev. 2020 Jun;43(3):903-910. doi: 10.1007/s10143-019-01087-3. Epub 2019 Feb 20. Neurosurg Rev. 2020. PMID: 30788677
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
