

The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia
- PMID: 36142738
- PMCID: PMC9502731
- DOI: 10.3390/ijms231810811
The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia
Abstract
β-Thalassemia is one of the most common genetically inherited disorders worldwide, and it is characterized by defective β-globin chain synthesis leading to reduced or absent β-globin chains. The excess α-globin chains are the key factor leading to the death of differentiating erythroblasts in a process termed ineffective erythropoiesis, leading to anemia and associated complications in patients. The mechanism of ineffective erythropoiesis in β-thalassemia is complex and not fully understood. Autophagy is primarily known as a cell recycling mechanism in which old or dysfunctional proteins and organelles are digested to allow recycling of constituent elements. In late stage, erythropoiesis autophagy is involved in the removal of mitochondria as part of terminal differentiation. Several studies have shown that autophagy is increased in earlier erythropoiesis in β-thalassemia erythroblasts, as compared to normal erythroblasts. This review summarizes what is known about the role of autophagy in β-thalassemia erythropoiesis and shows that modulation of autophagy and its interplay with apoptosis may provide a new therapeutic route in the treatment of β-thalassemia. Literature was searched and relevant articles were collected from databases, including PubMed, Scopus, Prospero, Clinicaltrials.gov, Google Scholar, and the Google search engine. Search terms included: β-thalassemia, ineffective erythropoiesis, autophagy, novel treatment, and drugs during the initial search. Relevant titles and abstracts were screened to choose relevant articles. Further, selected full-text articles were retrieved, and then, relevant cross-references were scanned to collect further information for the present review.
Keywords: ER stress; apoptosis; autophagy; ineffective erythropoiesis; β-thalassemia.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
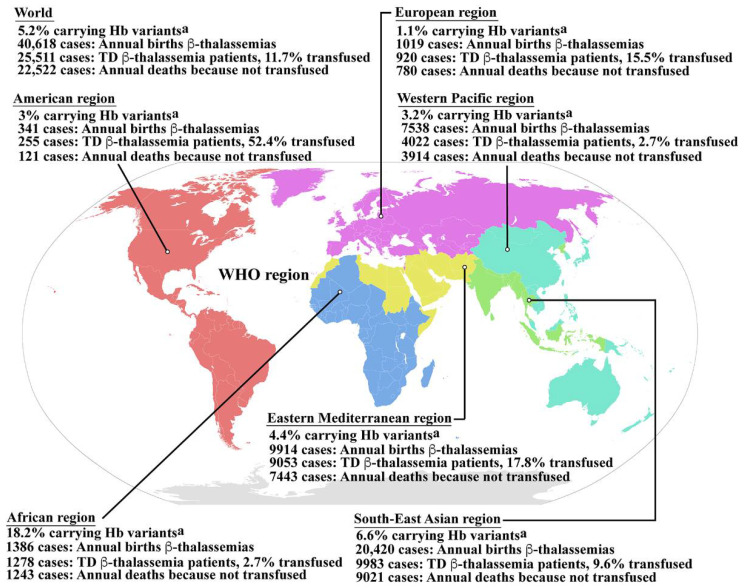
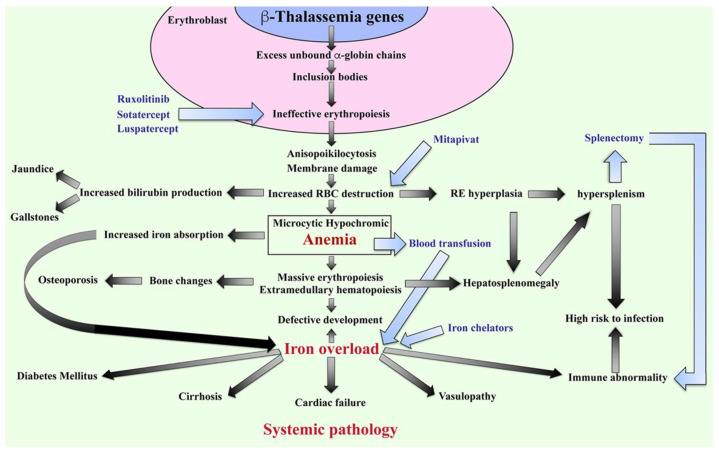
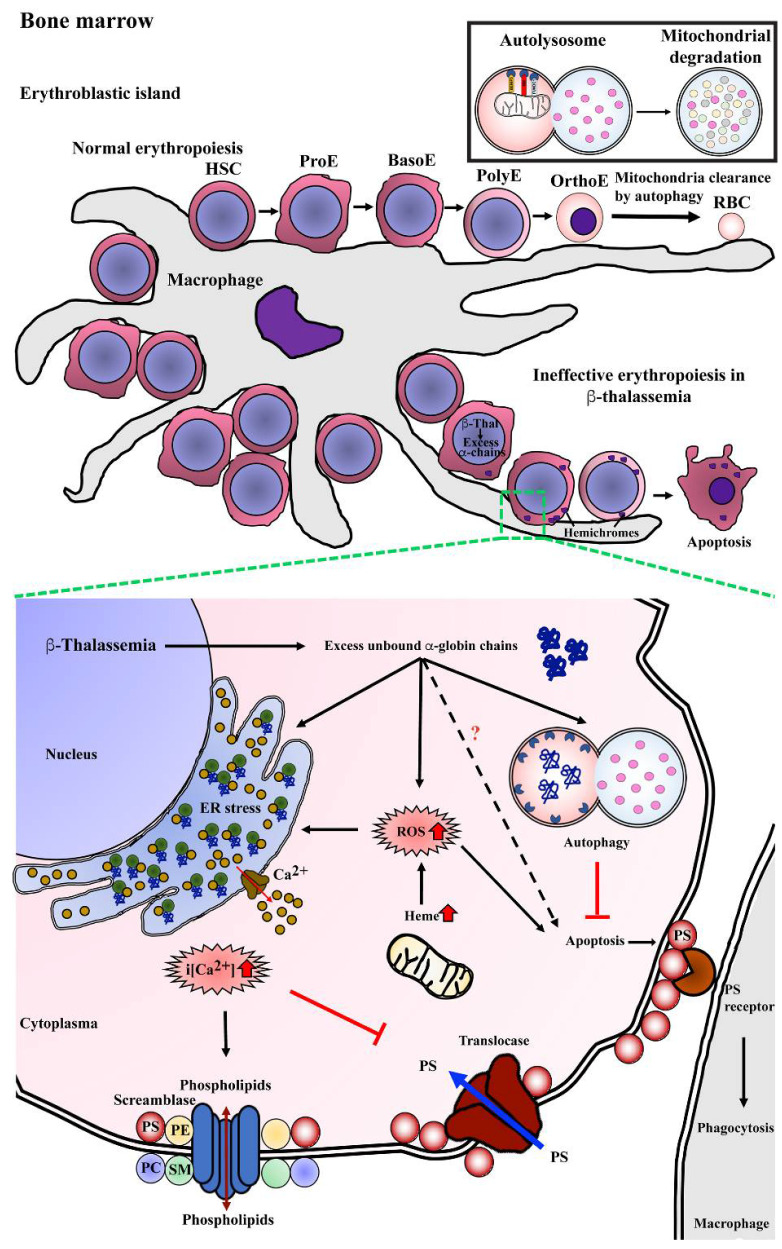
Similar articles
-
Improving Ineffective Erythropoiesis in Thalassemia: A Hope on the Horizon.Cureus. 2021 Oct 5;13(10):e18502. doi: 10.7759/cureus.18502. eCollection 2021 Oct. Cureus. 2021. PMID: 34754662 Free PMC article. Review.
-
Enhanced activation of autophagy in β-thalassemia/Hb E erythroblasts during erythropoiesis.Ann Hematol. 2011 Jul;90(7):747-58. doi: 10.1007/s00277-010-1152-5. Epub 2011 Jan 8. Ann Hematol. 2011. PMID: 21221583
-
Mitoxantrone ameliorates ineffective erythropoiesis in a β-thalassemia intermedia mouse model.Blood Adv. 2024 Aug 13;8(15):4017-4024. doi: 10.1182/bloodadvances.2024012679. Blood Adv. 2024. PMID: 38861356 Free PMC article.
-
HSP70 sequestration by free α-globin promotes ineffective erythropoiesis in β-thalassaemia.Nature. 2014 Oct 9;514(7521):242-6. doi: 10.1038/nature13614. Epub 2014 Aug 24. Nature. 2014. PMID: 25156257
-
Protein quality control during erythropoiesis and hemoglobin synthesis.Hematol Oncol Clin North Am. 2010 Dec;24(6):1071-88. doi: 10.1016/j.hoc.2010.08.013. Hematol Oncol Clin North Am. 2010. PMID: 21075281 Free PMC article. Review.
Cited by
-
Discovery-Based Proteomics Identify Skeletal Muscle Mitochondrial Alterations as an Early Metabolic Defect in a Mouse Model of β-Thalassemia.Int J Mol Sci. 2023 Feb 23;24(5):4402. doi: 10.3390/ijms24054402. Int J Mol Sci. 2023. PMID: 36901833 Free PMC article.
-
Are Mitochondria a Potential Target for Treating β-Thalassemia?J Clin Med. 2025 Feb 8;14(4):1095. doi: 10.3390/jcm14041095. J Clin Med. 2025. PMID: 40004626 Free PMC article. Review.
-
Therapeutic Relevance of Inducing Autophagy in β-Thalassemia.Cells. 2024 May 25;13(11):918. doi: 10.3390/cells13110918. Cells. 2024. PMID: 38891049 Free PMC article. Review.
-
Current understanding of eryptosis: mechanisms, physiological functions, role in disease, pharmacological applications, and nomenclature recommendations.Cell Death Dis. 2025 Jul 1;16(1):467. doi: 10.1038/s41419-025-07784-w. Cell Death Dis. 2025. PMID: 40592821 Free PMC article. Review.
-
Impact of α-Globin Gene Expression and α-Globin Modifiers on the Phenotype of β-Thalassemia and Other Hemoglobinopathies: Implications for Patient Management.Int J Mol Sci. 2024 Mar 17;25(6):3400. doi: 10.3390/ijms25063400. Int J Mol Sci. 2024. PMID: 38542374 Free PMC article. Review.
References
-
- Pootrakul P., Sirankapracha P., Hemsorach S., Moungsub W., Kumbunlue R., Piangitjagum A., Wasi P., Ma L., Schrier S.L. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in thai patients with thalassemia. Blood. 2000;96:2606–2612. doi: 10.1182/blood.V96.7.2606.h8002606_2606_2612. - DOI - PubMed
-
- Schweers R.L., Zhang J., Randall M.S., Loyd M.R., Li W., Dorsey F.C., Kundu M., Opferman J.T., Cleveland J.L., Miller J.L., et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
