

Past, Present, and Future of Genome Modification in Escherichia coli
- PMID: 36144436
- PMCID: PMC9504249
- DOI: 10.3390/microorganisms10091835
Past, Present, and Future of Genome Modification in Escherichia coli
Abstract
Escherichia coli K-12 is one of the most well-studied species of bacteria. This species, however, is much more difficult to modify by homologous recombination (HR) than other model microorganisms. Research on HR in E. coli has led to a better understanding of the molecular mechanisms of HR, resulting in technical improvements and rapid progress in genome research, and allowing whole-genome mutagenesis and large-scale genome modifications. Developments using λ Red (exo, bet, and gam) and CRISPR-Cas have made E. coli as amenable to genome modification as other model microorganisms, such as Saccharomyces cerevisiae and Bacillus subtilis. This review describes the history of recombination research in E. coli, as well as improvements in techniques for genome modification by HR. This review also describes the results of large-scale genome modification of E. coli using these technologies, including DNA synthesis and assembly. In addition, this article reviews recent advances in genome modification, considers future directions, and describes problems associated with the creation of cells by design.
Keywords: Escherichia coli K-12; HR; P1 transduction; genome modification; homologous recombination; mutation; recombineering; site-specific recombination; λ Red.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
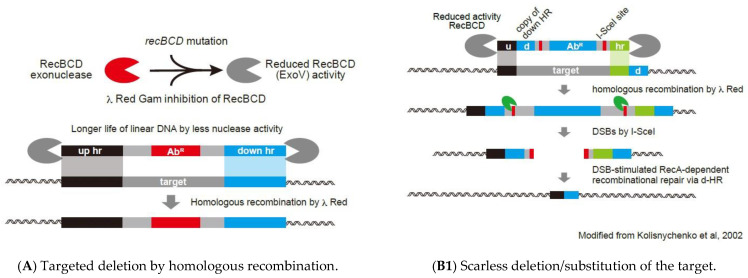
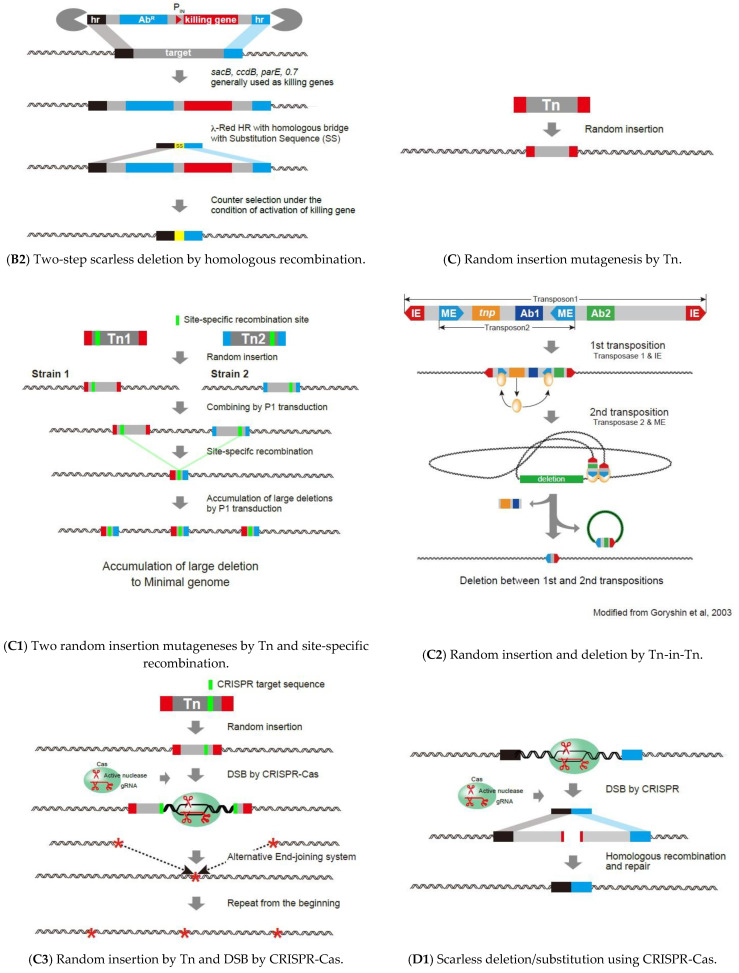
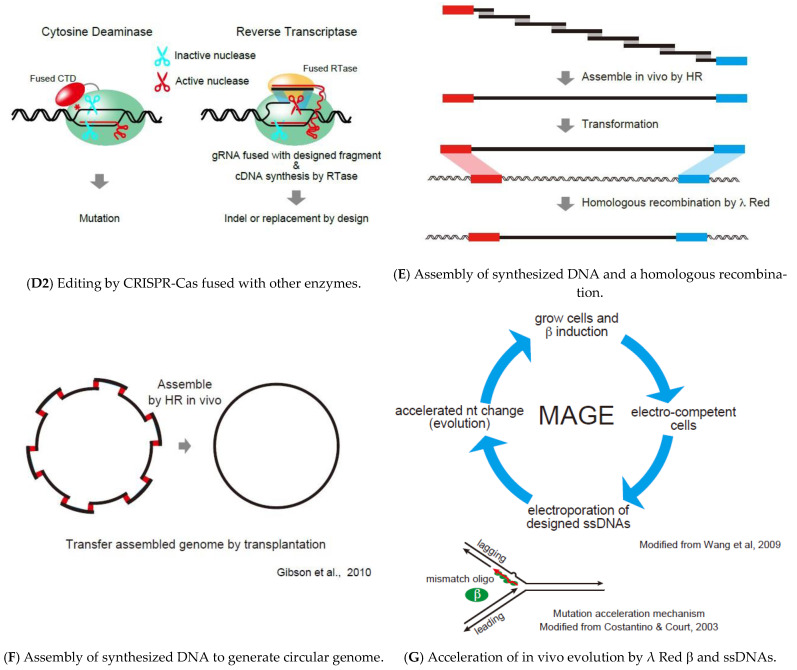
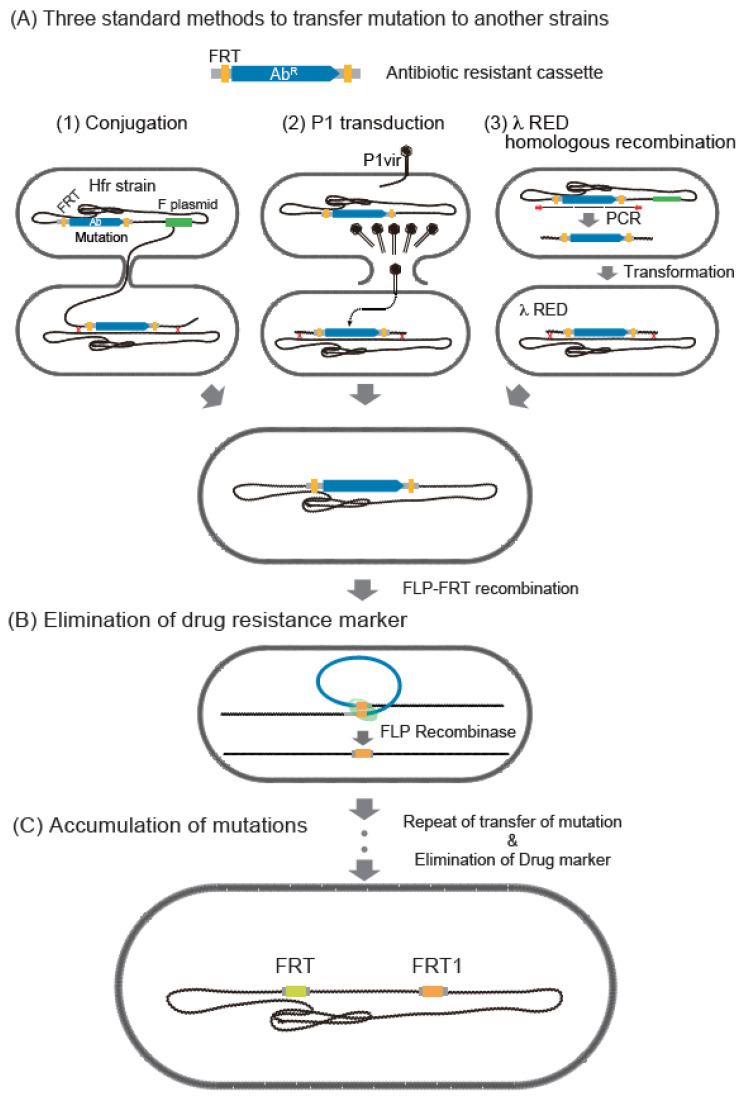
Similar articles
-
A double-locus scarless genome editing system in Escherichia coli.Biotechnol Lett. 2020 Aug;42(8):1457-1465. doi: 10.1007/s10529-020-02856-7. Epub 2020 Mar 4. Biotechnol Lett. 2020. PMID: 32130564
-
Viral recombination systems limit CRISPR-Cas targeting through the generation of escape mutations.Cell Host Microbe. 2021 Oct 13;29(10):1482-1495.e12. doi: 10.1016/j.chom.2021.09.001. Epub 2021 Sep 27. Cell Host Microbe. 2021. PMID: 34582782 Free PMC article.
-
Construction and functional characterization of an integrative form lambda Red recombineering Escherichia coli strain.FEMS Microbiol Lett. 2010 Aug 1;309(2):178-83. doi: 10.1111/j.1574-6968.2010.02036.x. Epub 2010 Jun 16. FEMS Microbiol Lett. 2010. PMID: 20618864
-
[Application of gene editing technology in Escherichia coli].Sheng Wu Gong Cheng Xue Bao. 2022 Apr 25;38(4):1446-1461. doi: 10.13345/j.cjb.210680. Sheng Wu Gong Cheng Xue Bao. 2022. PMID: 35470618 Review. Chinese.
-
Bacterial Recombineering: Genome Engineering via Phage-Based Homologous Recombination.ACS Synth Biol. 2015 Nov 20;4(11):1176-85. doi: 10.1021/acssynbio.5b00009. Epub 2015 Apr 27. ACS Synth Biol. 2015. PMID: 25856528 Review.
Cited by
-
Biocompatible Poly(ε-Caprolactone) Nanocapsules Enhance the Bioavailability, Antibacterial, and Immunomodulatory Activities of Curcumin.Int J Mol Sci. 2024 Oct 4;25(19):10692. doi: 10.3390/ijms251910692. Int J Mol Sci. 2024. PMID: 39409022 Free PMC article.
-
Regioselective O-acetylation of various glucosides catalyzed by Escherichia coli maltose O-acetyltransferase.Appl Microbiol Biotechnol. 2023 Dec;107(23):7031-7042. doi: 10.1007/s00253-023-12790-z. Epub 2023 Sep 20. Appl Microbiol Biotechnol. 2023. PMID: 37728626
-
Insights into the synthesis, engineering, and functions of microbial pigments in Deinococcus bacteria.Front Microbiol. 2024 Jul 25;15:1447785. doi: 10.3389/fmicb.2024.1447785. eCollection 2024. Front Microbiol. 2024. PMID: 39119139 Free PMC article. Review.
References
-
- Alexander E.H., Leidy G. Transformation of type specificity of Hemophilus influenzae. A.M.A. Am. J. Dis. Child. 1950;80:877–878. - PubMed
Publication types
Grants and funding
- 16H02485/Grant-in-Aid for Scientific Research (A), Ministry of Education, Culture, Sports, Science and Technology of Japan
- XTXM202202-2021A1515012401/Project of Collaborative Innovation Center of GDAAS,
- 2020B0202080004/Key Research and Development Program of Guangdong Province
- R2020PY-JC001/The special fund for scientific innovation strategy-construction of high-level Academy of Agriculture Science
- R2020YJ-LJ001/The special fund for scientific innovation strategy-construction of high-level Academy of Agriculture Science
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
