

Relative binding free energy calculations with transformato: A molecular dynamics engine-independent tool
- PMID: 36148009
- PMCID: PMC9485484
- DOI: 10.3389/fmolb.2022.954638
Relative binding free energy calculations with transformato: A molecular dynamics engine-independent tool
Abstract
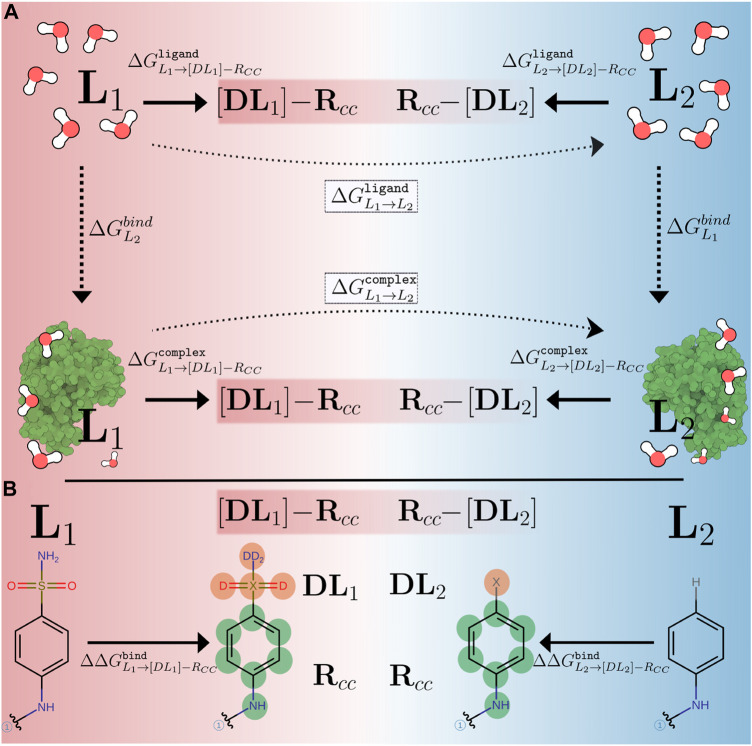
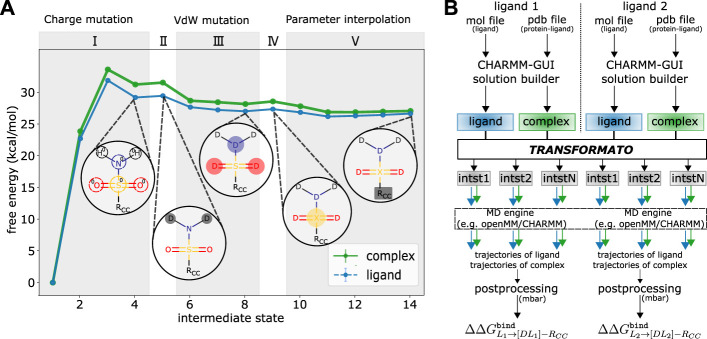
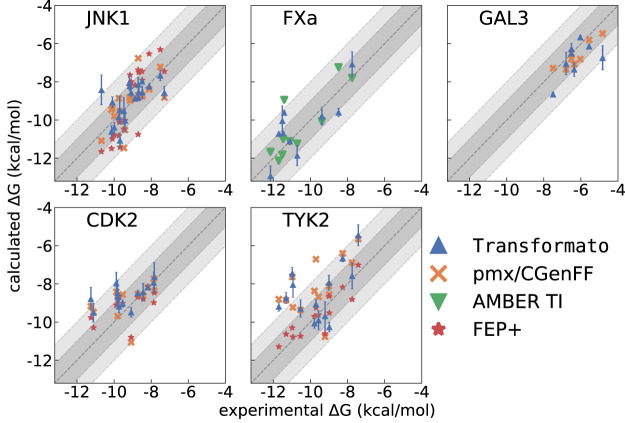
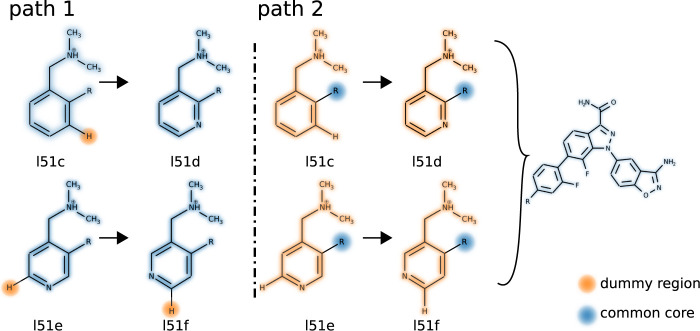
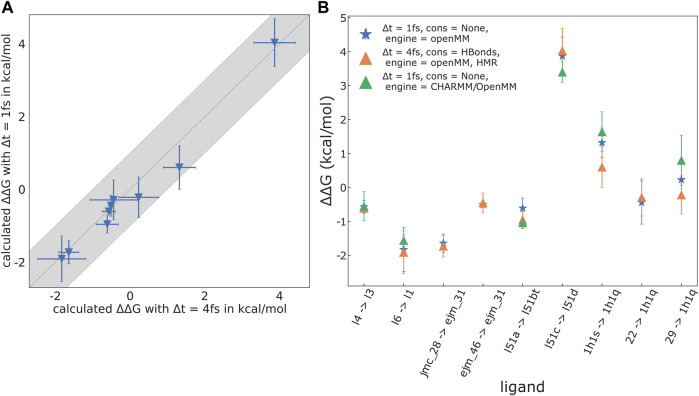
We present the software package transformato for the setup of large-scale relative binding free energy calculations. Transformato is written in Python as an open source project (https://github.com/wiederm/transformato); in contrast to comparable tools, it is not closely tied to a particular molecular dynamics engine to carry out the underlying simulations. Instead of alchemically transforming a ligand L 1 directly into another L 2, the two ligands are mutated to a common core. Thus, while dummy atoms are required at intermediate states, in particular at the common core state, none are present at the physical endstates. To validate the method, we calculated 76 relative binding free energy differences for five protein-ligand systems. The overall root mean squared error to experimental binding free energies is 1.17 kcal/mol with a Pearson correlation coefficient of 0.73. For selected cases, we checked that the relative binding free energy differences between pairs of ligands do not depend on the choice of the intermediate common core structure. Additionally, we report results with and without hydrogen mass reweighting. The code currently supports OpenMM, CHARMM, and CHARMM/OpenMM directly. Since the program logic to choose and construct alchemical transformation paths is separated from the generation of input and topology/parameter files, extending transformato to support additional molecular dynamics engines is straightforward.
Keywords: automated setup; binding affinity; free energy; molecular dynamics simulation; open source; python (programming language).
Copyright © 2022 Karwounopoulos, Wieder and Boresch.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Alchemical Free Energy Workflows for the Computation of Protein-Ligand Binding Affinities.Methods Mol Biol. 2024;2716:241-264. doi: 10.1007/978-1-0716-3449-3_11. Methods Mol Biol. 2024. PMID: 37702943
-
Alchemical free energy simulations without speed limits. A generic framework to calculate free energy differences independent of the underlying molecular dynamics program.J Comput Chem. 2022 Jun 30;43(17):1151-1160. doi: 10.1002/jcc.26877. Epub 2022 Apr 29. J Comput Chem. 2022. PMID: 35485139 Free PMC article.
-
A Python tool to set up relative free energy calculations in GROMACS.J Comput Aided Mol Des. 2015 Nov;29(11):1007-14. doi: 10.1007/s10822-015-9873-0. Epub 2015 Oct 20. J Comput Aided Mol Des. 2015. PMID: 26487189 Free PMC article.
-
CHARMM-GUI Free Energy Calculator for Practical Ligand Binding Free Energy Simulations with AMBER.J Chem Inf Model. 2021 Sep 27;61(9):4145-4151. doi: 10.1021/acs.jcim.1c00747. Epub 2021 Sep 15. J Chem Inf Model. 2021. PMID: 34521199 Free PMC article.
-
Tinker-OpenMM: Absolute and relative alchemical free energies using AMOEBA on GPUs.J Comput Chem. 2017 Sep 5;38(23):2047-2055. doi: 10.1002/jcc.24853. Epub 2017 Jun 10. J Comput Chem. 2017. PMID: 28600826 Free PMC article.
Cited by
-
Broadening the Scope of Binding Free Energy Calculations Using a Separated Topologies Approach.J Chem Theory Comput. 2023 Aug 8;19(15):5058-5076. doi: 10.1021/acs.jctc.3c00282. Epub 2023 Jul 24. J Chem Theory Comput. 2023. PMID: 37487138 Free PMC article.
-
Ligand Binding Affinity Prediction for Membrane Proteins with Alchemical Free Energy Calculation Methods.J Chem Inf Model. 2024 Jul 22;64(14):5671-5679. doi: 10.1021/acs.jcim.4c00764. Epub 2024 Jul 3. J Chem Inf Model. 2024. PMID: 38959405 Free PMC article.
-
Alchemical Free Energy Workflows for the Computation of Protein-Ligand Binding Affinities.Methods Mol Biol. 2024;2716:241-264. doi: 10.1007/978-1-0716-3449-3_11. Methods Mol Biol. 2024. PMID: 37702943
-
Updates to Protex for Simulating Proton Transfers in an Ionic Liquid.J Phys Chem B. 2024 Apr 11;128(14):3416-3426. doi: 10.1021/acs.jpcb.3c07356. Epub 2024 Apr 1. J Phys Chem B. 2024. PMID: 38557106 Free PMC article.
-
Insights and Challenges in Correcting Force Field Based Solvation Free Energies Using a Neural Network Potential.J Phys Chem B. 2024 Jul 18;128(28):6693-6703. doi: 10.1021/acs.jpcb.4c01417. Epub 2024 Jul 8. J Phys Chem B. 2024. PMID: 38976601 Free PMC article.
References
-
- Åqvist J., Wennerström P., Nervall M., Bjelic S., Brandsdal B. O. (2004). Molecular dynamics simulations of water and biomolecules with a Monte Carlo constant pressure algorithm. Chem. Phys. Lett. 384, 288–294. 10.1016/J.CPLETT.2003.12.039 - DOI
-
- Boresch S., Karplus M. (1998). The role of bonded terms in free energy simulations: 1. theoretical analysis. J. Phys. Chem. A 103, 103–118. 10.1021/jp981628n - DOI
-
- Chow K. H., Ferguson D. M. (1995). Isothermal-isobaric molecular dynamics simulations with Monte Carlo volume sampling. Comput. Phys. Commun. 91, 283–289. 10.1016/0010-4655(95)00059-O - DOI
LinkOut - more resources
Full Text Sources