

Deep mutational learning predicts ACE2 binding and antibody escape to combinatorial mutations in the SARS-CoV-2 receptor-binding domain
- PMID: 36150393
- PMCID: PMC9428596
- DOI: 10.1016/j.cell.2022.08.024
Deep mutational learning predicts ACE2 binding and antibody escape to combinatorial mutations in the SARS-CoV-2 receptor-binding domain
Abstract
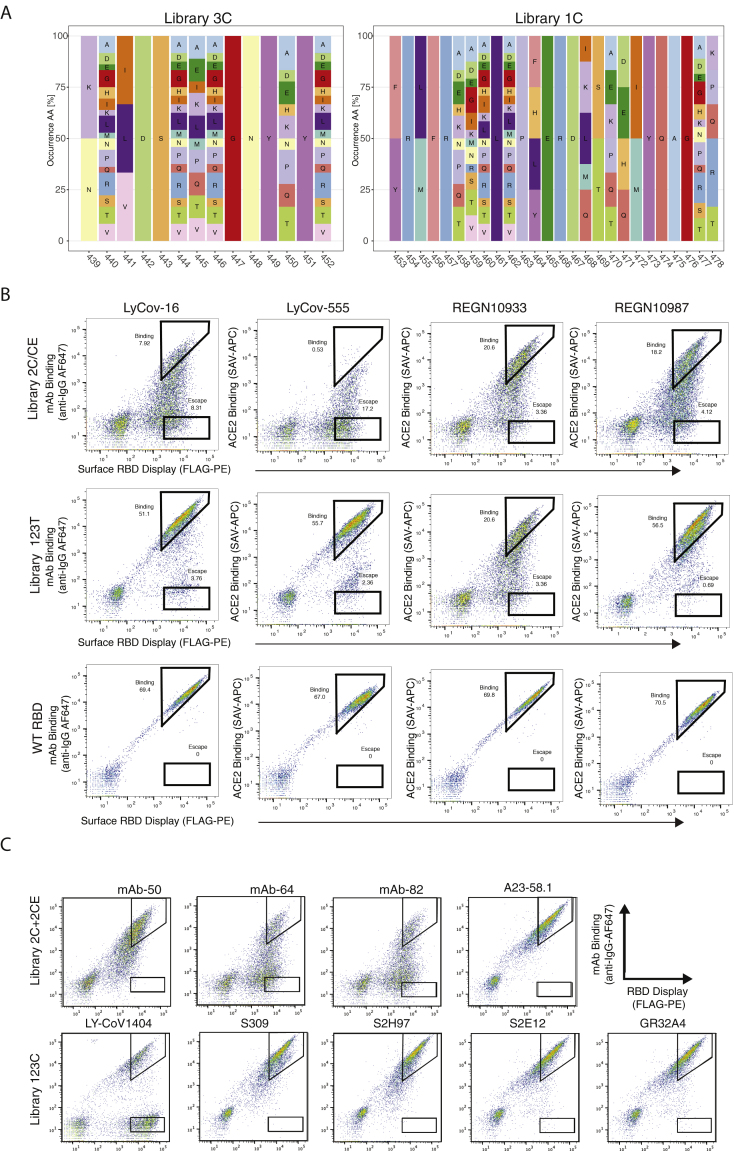
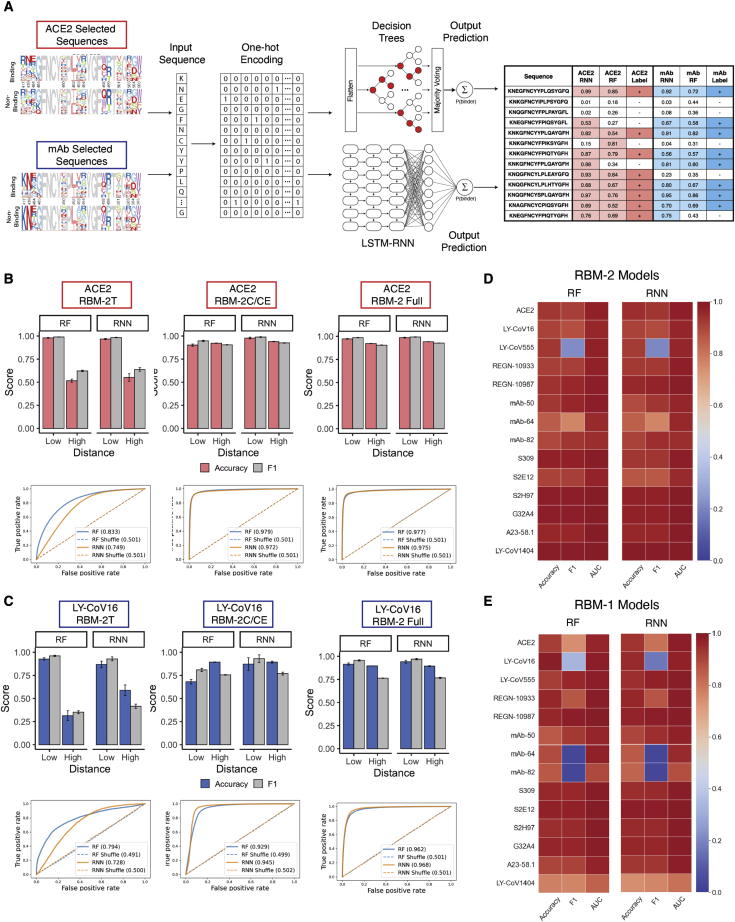
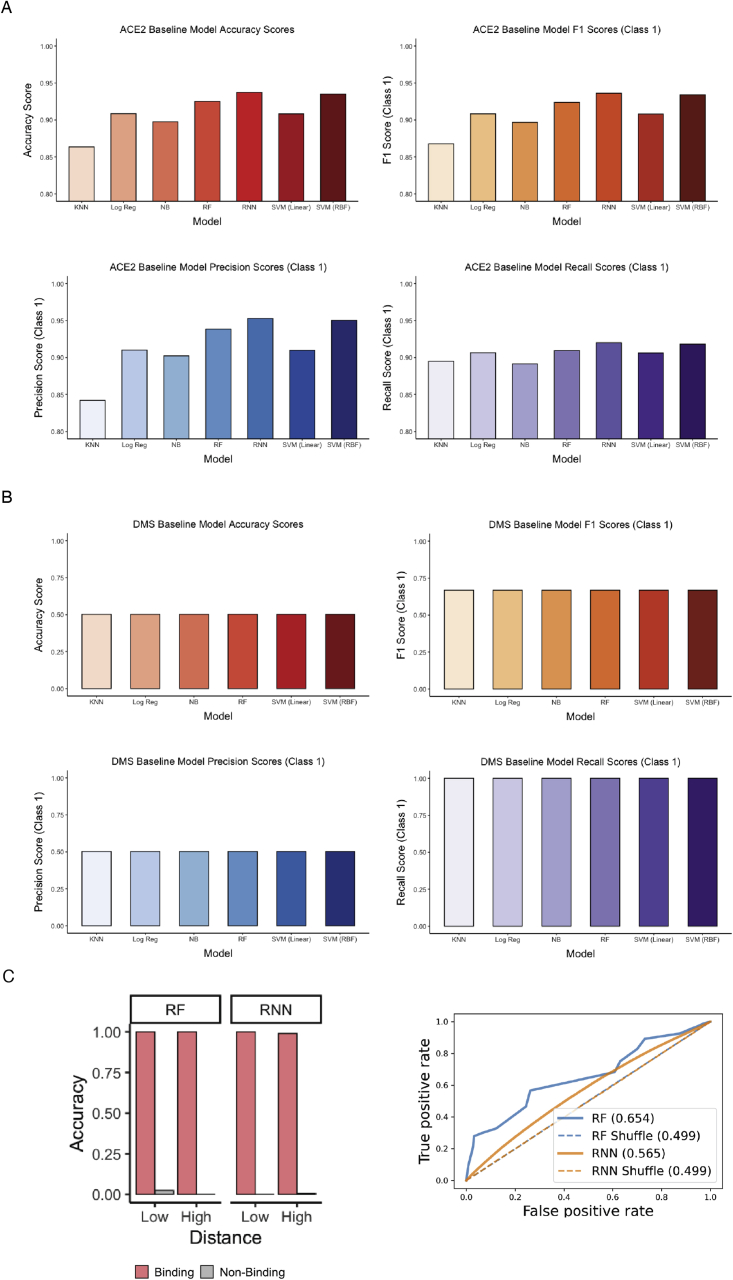
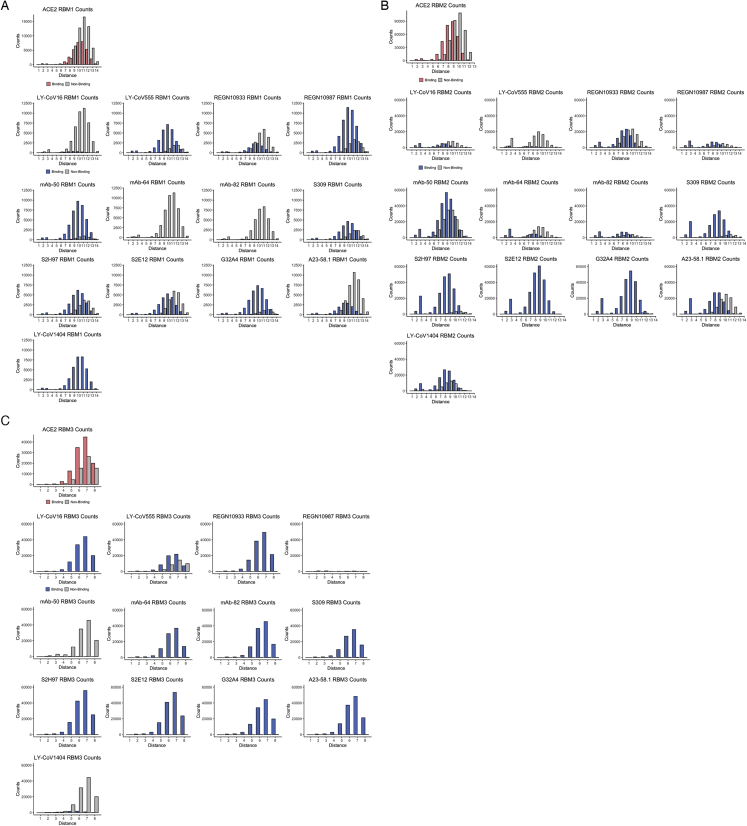
The continual evolution of SARS-CoV-2 and the emergence of variants that show resistance to vaccines and neutralizing antibodies threaten to prolong the COVID-19 pandemic. Selection and emergence of SARS-CoV-2 variants are driven in part by mutations within the viral spike protein and in particular the ACE2 receptor-binding domain (RBD), a primary target site for neutralizing antibodies. Here, we develop deep mutational learning (DML), a machine-learning-guided protein engineering technology, which is used to investigate a massive sequence space of combinatorial mutations, representing billions of RBD variants, by accurately predicting their impact on ACE2 binding and antibody escape. A highly diverse landscape of possible SARS-CoV-2 variants is identified that could emerge from a multitude of evolutionary trajectories. DML may be used for predictive profiling on current and prospective variants, including highly mutated variants such as Omicron, thus guiding the development of therapeutic antibody treatments and vaccines for COVID-19.
Keywords: artificial intelligence; deep learning; deep sequencing; directed evolution; machine learning; protein engineering; viral escape; yeast display.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests ETH Zurich has filed for patent protection on the technology described herein, and J.M.T., C.R.W., B.G., R.A.E., and S.T.R. are named as co-inventors. C.R.W. is an employee of Alloy Therapeutics (Switzerland) AG. C.R.W. and S.T.R. may hold shares of Alloy Therapeutics. S.T.R. is on the scientific advisory board of Alloy Therapeutics.
Figures

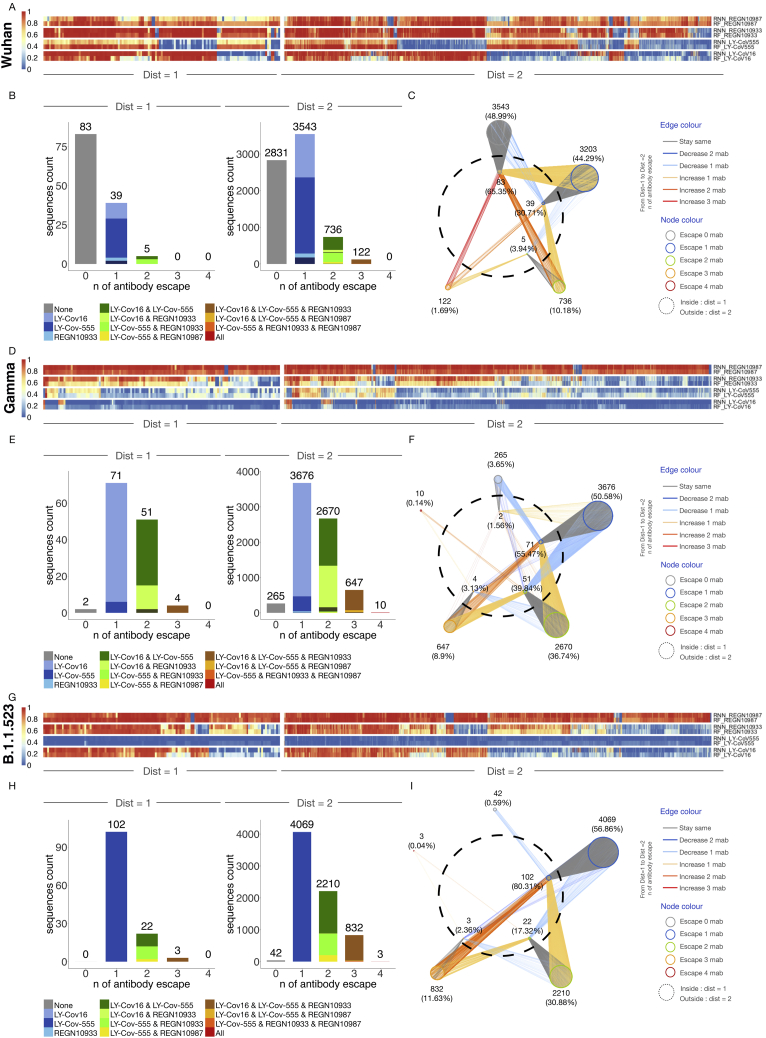
References
-
- Akbar R., Robert P.A., Weber C.R., Widrich M., Frank R., Pavlović M., Scheffer L., Chernigovskaya M., Snapkov I., Slabodkin A., et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. bioRxiv. 2021 doi: 10.1101/2021.07.08.451480. Preprint at. - DOI - PMC - PubMed
-
- Barnes C.O., West A.P., Huey-Tubman K.E., Hoffmann M.A.G., Sharaf N.G., Hoffman P.R., Koranda N., Gristick H.B., Gaebler C., Muecksch F., et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell. 2020;182:828–842.e16. doi: 10.1016/j.cell.2020.06.025. - DOI - PMC - PubMed
-
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014–1018. doi: 10.1126/science.abd0831. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
