

Genomic and biological study of fusion genes as resistance mechanisms to EGFR inhibitors
- PMID: 36153311
- PMCID: PMC9509394
- DOI: 10.1038/s41467-022-33210-2
Genomic and biological study of fusion genes as resistance mechanisms to EGFR inhibitors
Abstract
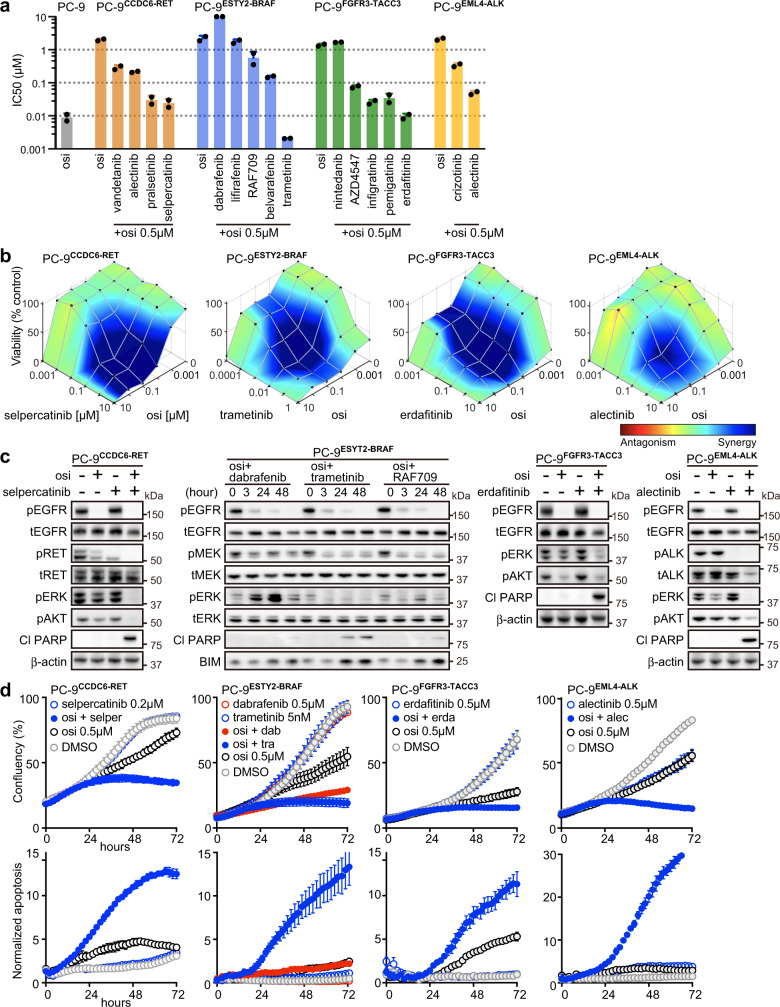
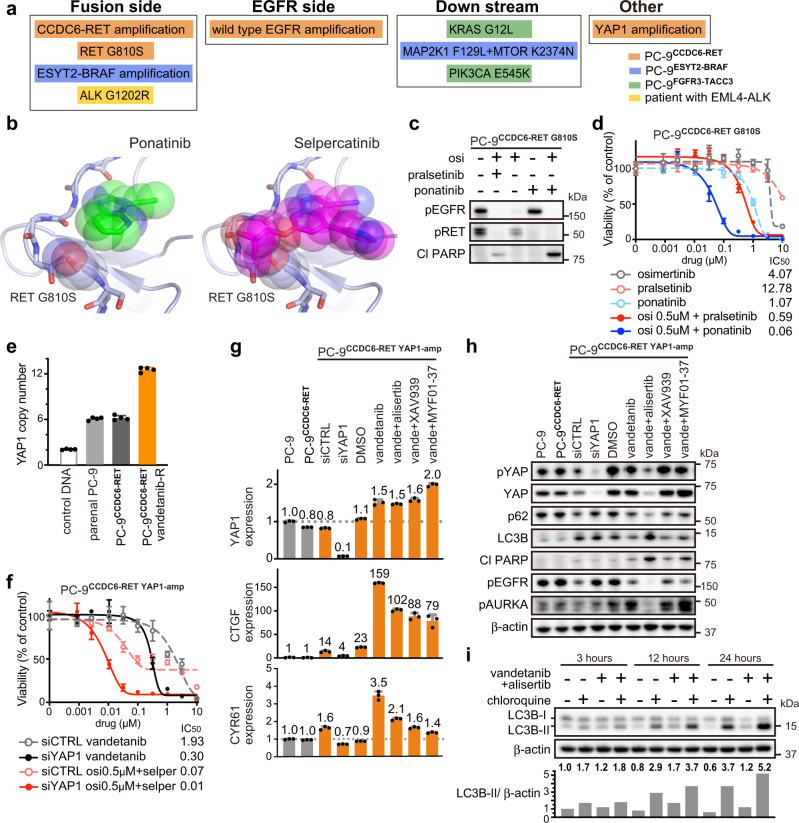
The clinical significance of gene fusions detected by DNA-based next generation sequencing remains unclear as resistance mechanisms to EGFR tyrosine kinase inhibitors in EGFR mutant non-small cell lung cancer. By studying EGFR inhibitor-resistant patients treated with a combination of an EGFR inhibitor and a drug targeting the putative resistance-causing fusion oncogene, we identify patients who benefit and those who do not from this treatment approach. Through evaluation including RNA-seq of potential drug resistance-imparting fusion oncogenes in 504 patients with EGFR mutant lung cancer, we identify only a minority of them as functional, potentially capable of imparting EGFR inhibitor resistance. We further functionally validate fusion oncogenes in vitro using CRISPR-based editing of EGFR mutant cell lines and use these models to identify known and unknown drug resistance mechanisms to combination therapies. Collectively, our results partially reveal the complex nature of fusion oncogenes as potential drug resistance mechanisms and highlight approaches that can be undertaken to determine their functional significance.
© 2022. The Author(s).
Conflict of interest statement
G.R.O. reports employment with foundation medicine and equity in Roche. D.E.H. reports consultant fees from Logos Capital. K.P.S. reports consultant fees from Eurofarma. L.M.S. reports consultant fees from EMD Serono, research grant from Genentech, and stock ownership in Moderna. P.A.J. reports consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, Loxo Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Silicon Therapeutics, Nuvalent, Esai, Bayer, and Biocartis; receiving post-marketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp; sponsored research agreements with AstraZeneca, Daichi-Sankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines and Astellas Pharmaceuticals; and stock ownership in Loxo Oncology and Gatekeeper Pharmaceuticals. All other authors report no conflicts of interest.
Figures
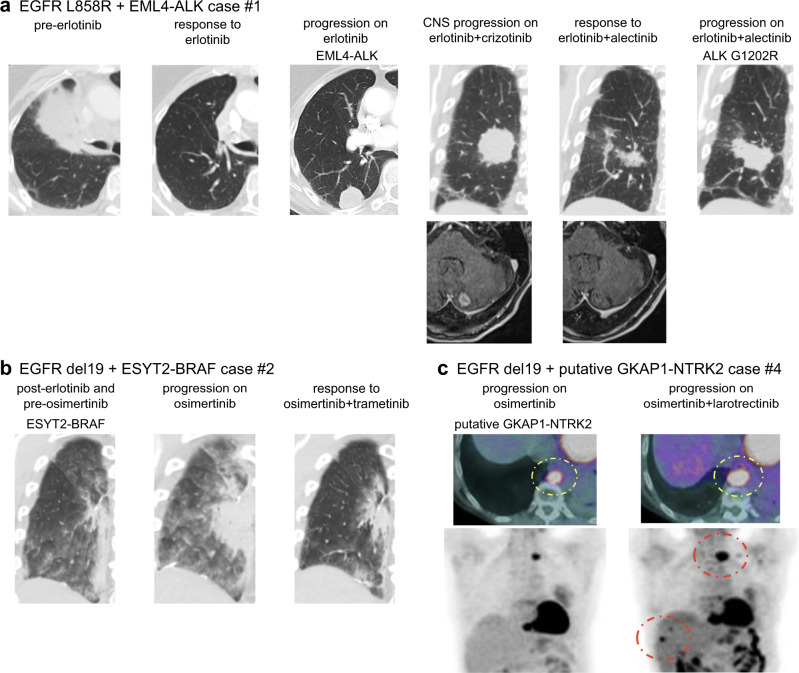

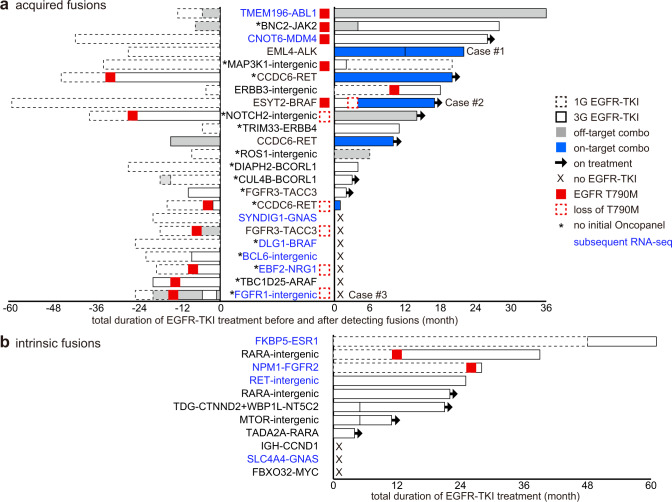
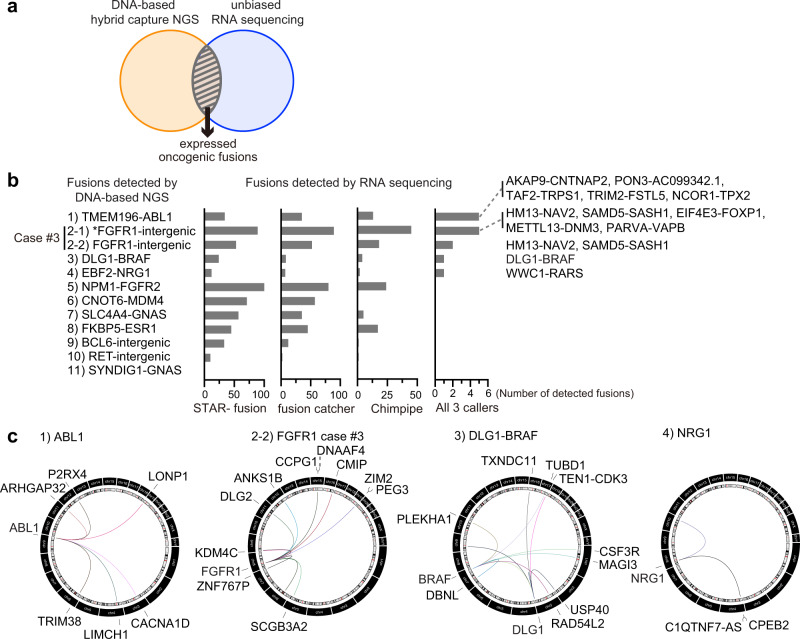
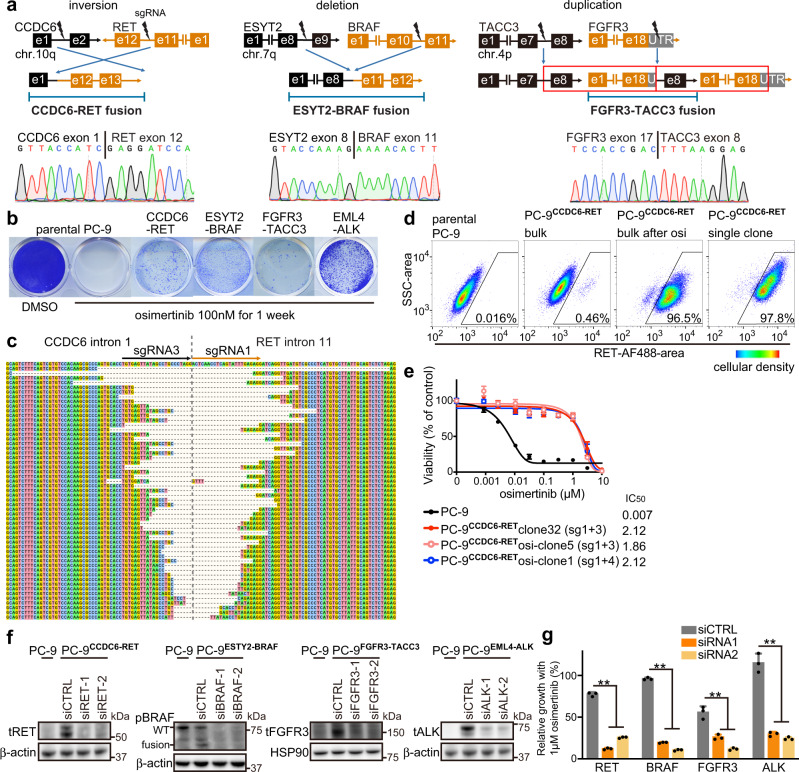
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
