

Mitochondrial protein dysfunction in pathogenesis of neurological diseases
- PMID: 36157077
- PMCID: PMC9489860
- DOI: 10.3389/fnmol.2022.974480
Mitochondrial protein dysfunction in pathogenesis of neurological diseases
Abstract
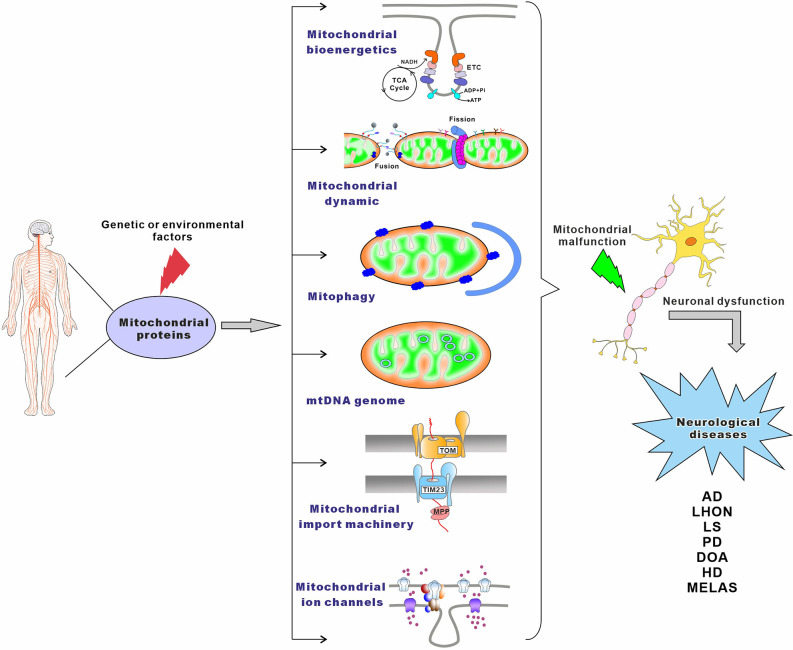
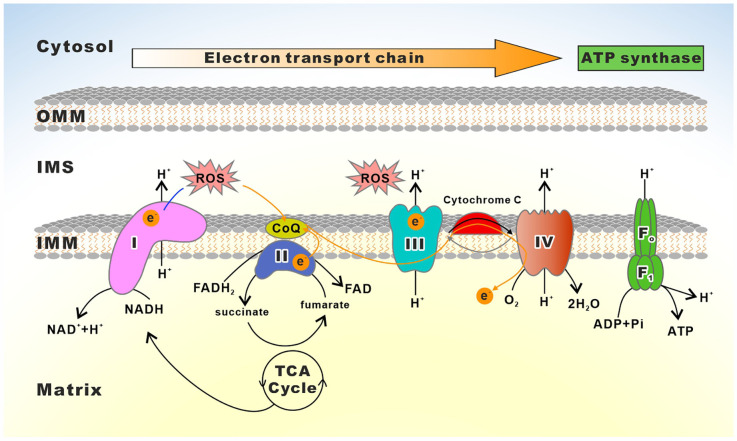
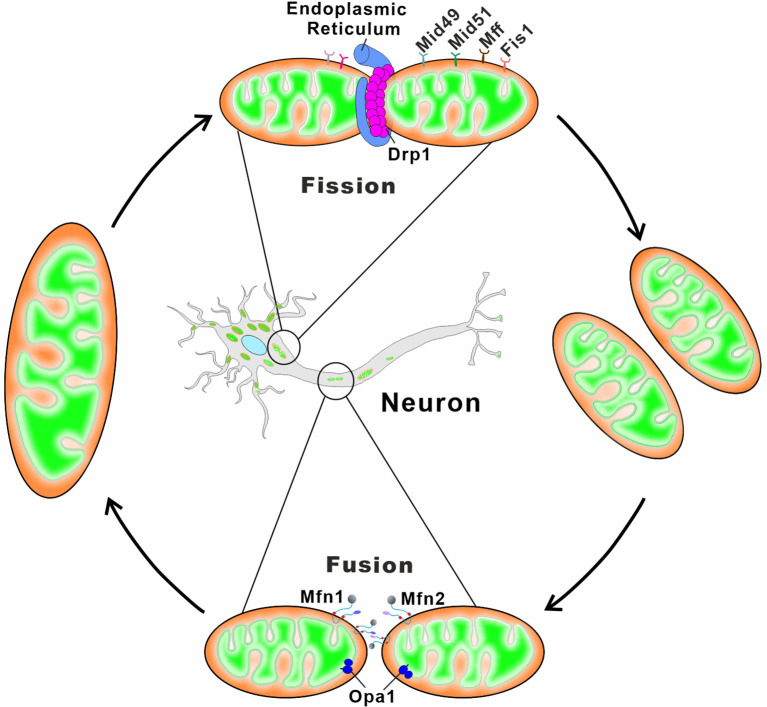
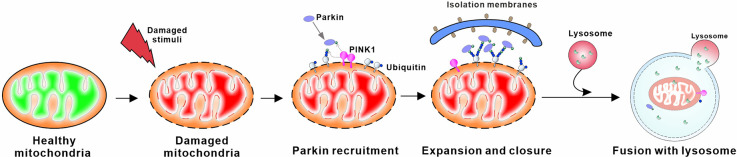
Mitochondria are essential organelles for neuronal function and cell survival. Besides the well-known bioenergetics, additional mitochondrial roles in calcium signaling, lipid biogenesis, regulation of reactive oxygen species, and apoptosis are pivotal in diverse cellular processes. The mitochondrial proteome encompasses about 1,500 proteins encoded by both the nuclear DNA and the maternally inherited mitochondrial DNA. Mutations in the nuclear or mitochondrial genome, or combinations of both, can result in mitochondrial protein deficiencies and mitochondrial malfunction. Therefore, mitochondrial quality control by proteins involved in various surveillance mechanisms is critical for neuronal integrity and viability. Abnormal proteins involved in mitochondrial bioenergetics, dynamics, mitophagy, import machinery, ion channels, and mitochondrial DNA maintenance have been linked to the pathogenesis of a number of neurological diseases. The goal of this review is to give an overview of these pathways and to summarize the interconnections between mitochondrial protein dysfunction and neurological diseases.
Keywords: mitochondrial bioenergetics; mitochondrial dynamics; mitochondrial import machinery; mitochondrial proteins; mitophagy; mtDNA maintenance; neurological diseases; pathogenesis.
Copyright © 2022 Wang, Yang, He, Pu, Yang, Wu, Zhou, Cen and Zhao.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Mitochondrial biogenesis: pharmacological approaches.Curr Pharm Des. 2014;20(35):5507-9. doi: 10.2174/138161282035140911142118. Curr Pharm Des. 2014. PMID: 24606795
-
[Pathways for maintenance of mitochondrial DNA integrity and mitochondrial functions in cells exposed to ionizing radiation].Radiats Biol Radioecol. 2013 Mar-Apr;53(2):117-36. doi: 10.7868/s0869803113020045. Radiats Biol Radioecol. 2013. PMID: 23786028 Review. Russian.
-
Functional Interplay between Cristae Biogenesis, Mitochondrial Dynamics and Mitochondrial DNA Integrity.Int J Mol Sci. 2019 Sep 3;20(17):4311. doi: 10.3390/ijms20174311. Int J Mol Sci. 2019. PMID: 31484398 Free PMC article. Review.
-
Mitochondrial dynamics, cell death and the pathogenesis of Parkinson's disease.Apoptosis. 2010 Nov;15(11):1336-53. doi: 10.1007/s10495-010-0465-0. Apoptosis. 2010. PMID: 20131004 Review.
-
Mitochondrial Protein Import Dysfunction in Pathogenesis of Neurodegenerative Diseases.Mol Neurobiol. 2021 Apr;58(4):1418-1437. doi: 10.1007/s12035-020-02200-0. Epub 2020 Nov 12. Mol Neurobiol. 2021. PMID: 33180216 Review.
Cited by
-
GTPBP8 modulates mitochondrial fission through a Drp1-dependent process.J Cell Sci. 2024 Apr 15;137(8):jcs261612. doi: 10.1242/jcs.261612. Epub 2024 Apr 30. J Cell Sci. 2024. PMID: 38587461 Free PMC article.
-
Exploring the role of mitochondrial dysfunction and aging in COVID-19-Related neurological complications.Mol Biol Rep. 2025 May 21;52(1):479. doi: 10.1007/s11033-025-10586-0. Mol Biol Rep. 2025. PMID: 40397294 Review.
-
Crosstalk among mitophagy, pyroptosis, ferroptosis, and necroptosis in central nervous system injuries.Neural Regen Res. 2024 Aug 1;19(8):1660-1670. doi: 10.4103/1673-5374.389361. Epub 2023 Nov 8. Neural Regen Res. 2024. PMID: 38103229 Free PMC article.
-
Nitric Oxide Signaling and Sensing in Age-Related Diseases.Antioxidants (Basel). 2024 Oct 9;13(10):1213. doi: 10.3390/antiox13101213. Antioxidants (Basel). 2024. PMID: 39456466 Free PMC article. Review.
-
Nanotherapeutic and Nano-Bio Interface for Regeneration and Healing.Biomedicines. 2024 Dec 23;12(12):2927. doi: 10.3390/biomedicines12122927. Biomedicines. 2024. PMID: 39767834 Free PMC article. Review.
References
-
- Ababneh N. A., Barham R., Al-kurdi B., Ali D., Al S., Ismail M. A., et al. (2022). Generation of a human induced pluripotent stem cell (iPSC) line (JUCTCi019-A) from a patient with Charcot-Marie-Tooth disease type 2A2 (CMT2A2) due to a heterozygous missense substitution c.2119C>T (p.Arg707Trp) in MFN2 gene. Stem Cell Res. 62:102786. 10.1016/j.scr.2022.102786 - DOI - PubMed
-
- Abdoh R. M. (2019). Homozygote c. 1789c>t (p.arg597trp) polg gene mutation in consanguineous moroccan patient with predominant motor phenotype of sando syndrome. J. Neurol. Sci. 405:65. 10.1016/j.jns.2019.10.1684 - DOI
-
- Abdulhag U. N., Soiferman D., Schueler-Furman O., Miller C., Shaag A., Elpeleg O., et al. (2015). Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 23, 159–164. 10.1038/ejhg.2014.85 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources