

Novel compound heterozygous mutation of SLC12A3 in Gitelman syndrome co-existent with hyperthyroidism: A case report and literature review
- PMID: 36158002
- PMCID: PMC9353888
- DOI: 10.12998/wjcc.v10.i21.7483
Novel compound heterozygous mutation of SLC12A3 in Gitelman syndrome co-existent with hyperthyroidism: A case report and literature review
Abstract
Background: Gitelman syndrome (GS) is a rare inherited autosomal recessive tubulopathy, characterized clinically by hypokalemia, hypomagnesemia, hypocalciuria, and metabolic alkalosis, and is caused by an inactivating mutation in SLC12A3. GS is prone to misdiagnosis when occurring simultaneously with hyperthyroidism. It is important to consider the possibility of other diseases when hyperthyroidism is combined with hypokalemia, which is difficult to correct.
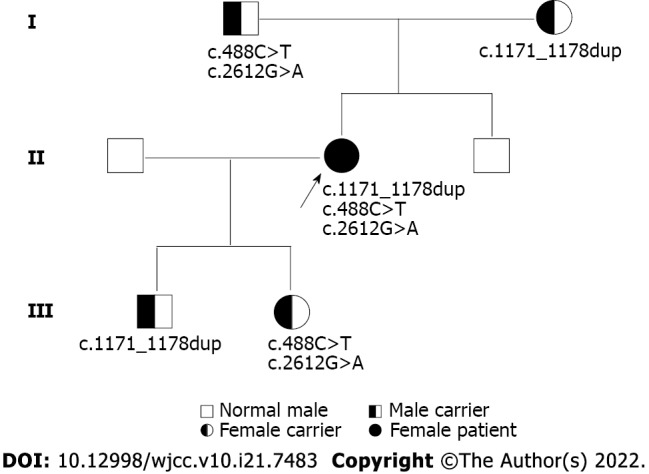
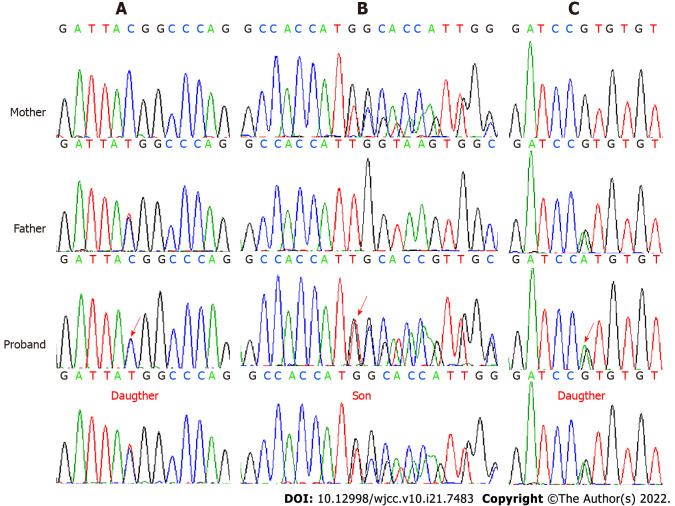
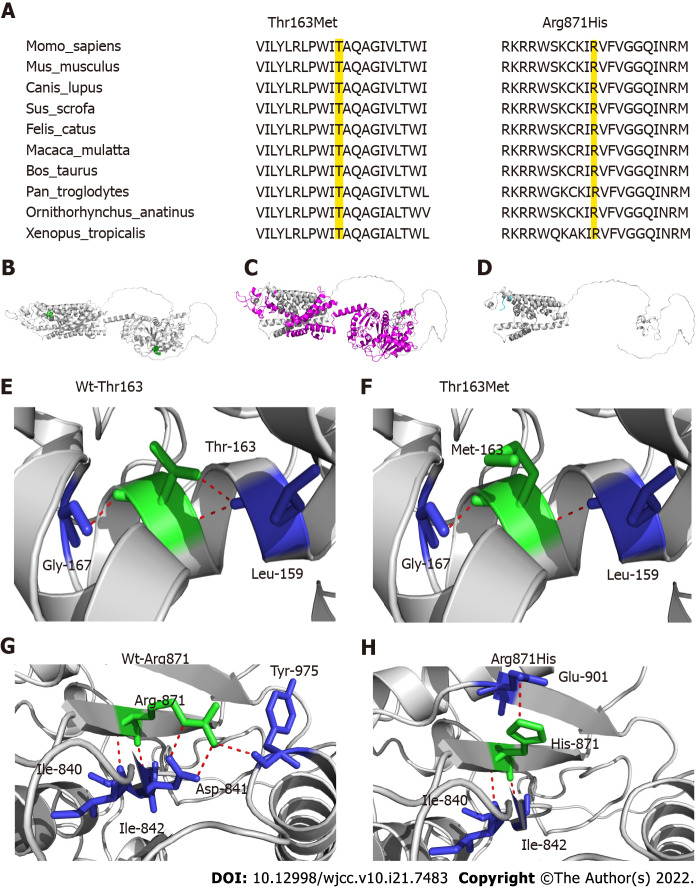
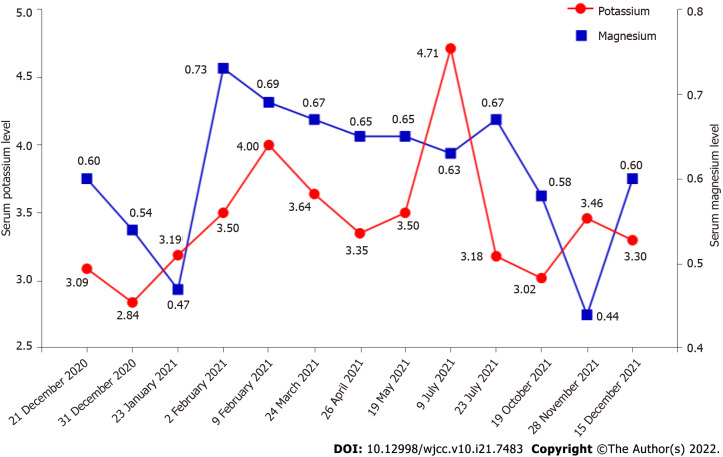
Case summary: A female patient with hyperthyroidism complicated with limb weakness was diagnosed with thyrotoxic hypokalemic periodic paralysis for 4 mo. However, the patient's serum potassium level remained low despite sufficient potassium replacement and remission of hyperthyroidism. GS was confirmed by whole exome and Sanger sequencing. Gene sequencing revealed compound heterozygous mutations of c.488C>T (p.Thr163Met), c.2612G>A (p.Arg871His), and c.1171_1178dupGCCACCAT (p.Ile393fs) in SLC12A3. Protein molecular modeling was performed to predict the effects of the identified missense mutations. All three mutations cause changes in protein structure and may result in abnormal protein function. All previously reported cases of GS coexisting with autoimmune thyroid disease are reviewed.
Conclusion: We have identified a novel compound heterozygous mutation in SLC12A3. The present study provides new genetic evidence for GS.
Keywords: Case report; Gene sequencing; Gitelman syndrome; Hyperthyroidism; Hypokalemia; SLC12A3.
©The Author(s) 2022. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Figures
References
-
- Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235. - PubMed
-
- Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12:24–30. - PubMed
-
- Abbasi B, Sharif Z, Sprabery LR. Hypokalemic thyrotoxic periodic paralysis with thyrotoxic psychosis and hypercapnic respiratory failure. Am J Med Sci. 2010;340:147–153. - PubMed
-
- Frese E, Brown M, Norton BJ. Clinical reliability of manual muscle testing. Middle trapezius and gluteus medius muscles. Phys Ther. 1987;67:1072–1076. - PubMed
-
- Zelikovic I, Szargel R, Hawash A, Labay V, Hatib I, Cohen N, Nakhoul F. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 2003;63:24–32. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
