Evolution of the ancestral mammalian karyotype and syntenic regions
- PMID: 36161960
- PMCID: PMC9550189
- DOI: 10.1073/pnas.2209139119
Evolution of the ancestral mammalian karyotype and syntenic regions
Abstract
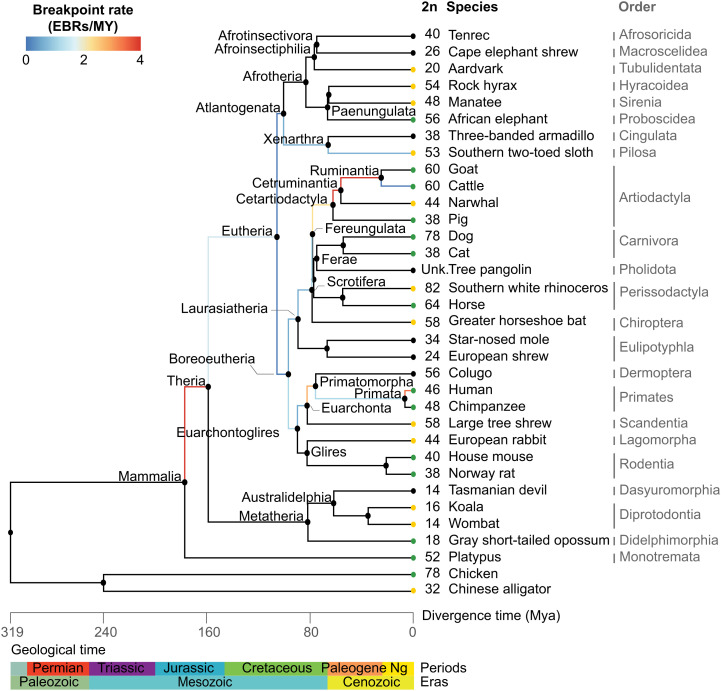
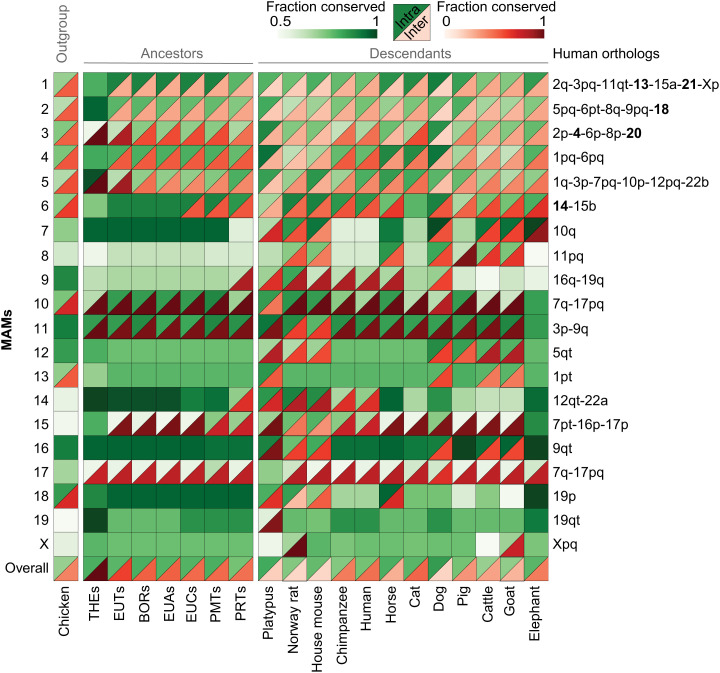
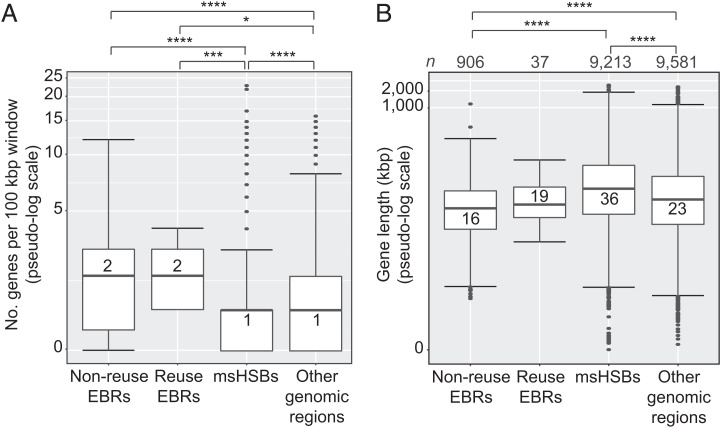
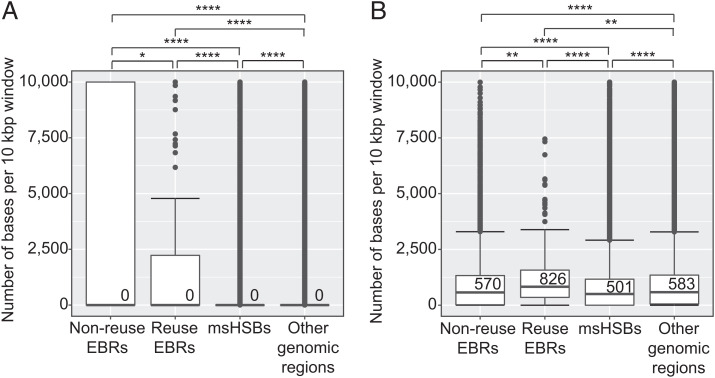
Decrypting the rearrangements that drive mammalian chromosome evolution is critical to understanding the molecular bases of speciation, adaptation, and disease susceptibility. Using 8 scaffolded and 26 chromosome-scale genome assemblies representing 23/26 mammal orders, we computationally reconstructed ancestral karyotypes and syntenic relationships at 16 nodes along the mammalian phylogeny. Three different reference genomes (human, sloth, and cattle) representing phylogenetically distinct mammalian superorders were used to assess reference bias in the reconstructed ancestral karyotypes and to expand the number of clades with reconstructed genomes. The mammalian ancestor likely had 19 pairs of autosomes, with nine of the smallest chromosomes shared with the common ancestor of all amniotes (three still conserved in extant mammals), demonstrating a striking conservation of synteny for ∼320 My of vertebrate evolution. The numbers and types of chromosome rearrangements were classified for transitions between the ancestral mammalian karyotype, descendent ancestors, and extant species. For example, 94 inversions, 16 fissions, and 14 fusions that occurred over 53 My differentiated the therian from the descendent eutherian ancestor. The highest breakpoint rate was observed between the mammalian and therian ancestors (3.9 breakpoints/My). Reconstructed mammalian ancestor chromosomes were found to have distinct evolutionary histories reflected in their rates and types of rearrangements. The distributions of genes, repetitive elements, topologically associating domains, and actively transcribed regions in multispecies homologous synteny blocks and evolutionary breakpoint regions indicate that purifying selection acted over millions of years of vertebrate evolution to maintain syntenic relationships of developmentally important genes and regulatory landscapes of gene-dense chromosomes.
Keywords: ancestral genome reconstruction; chromosome evolution; mammals; synteny conservation; topologically associating domains.
Conflict of interest statement
The authors declare no competing interest.
Figures