

Single-cell genome sequencing of human neurons identifies somatic point mutation and indel enrichment in regulatory elements
- PMID: 36163278
- PMCID: PMC9833626
- DOI: 10.1038/s41588-022-01180-2
Single-cell genome sequencing of human neurons identifies somatic point mutation and indel enrichment in regulatory elements
Abstract
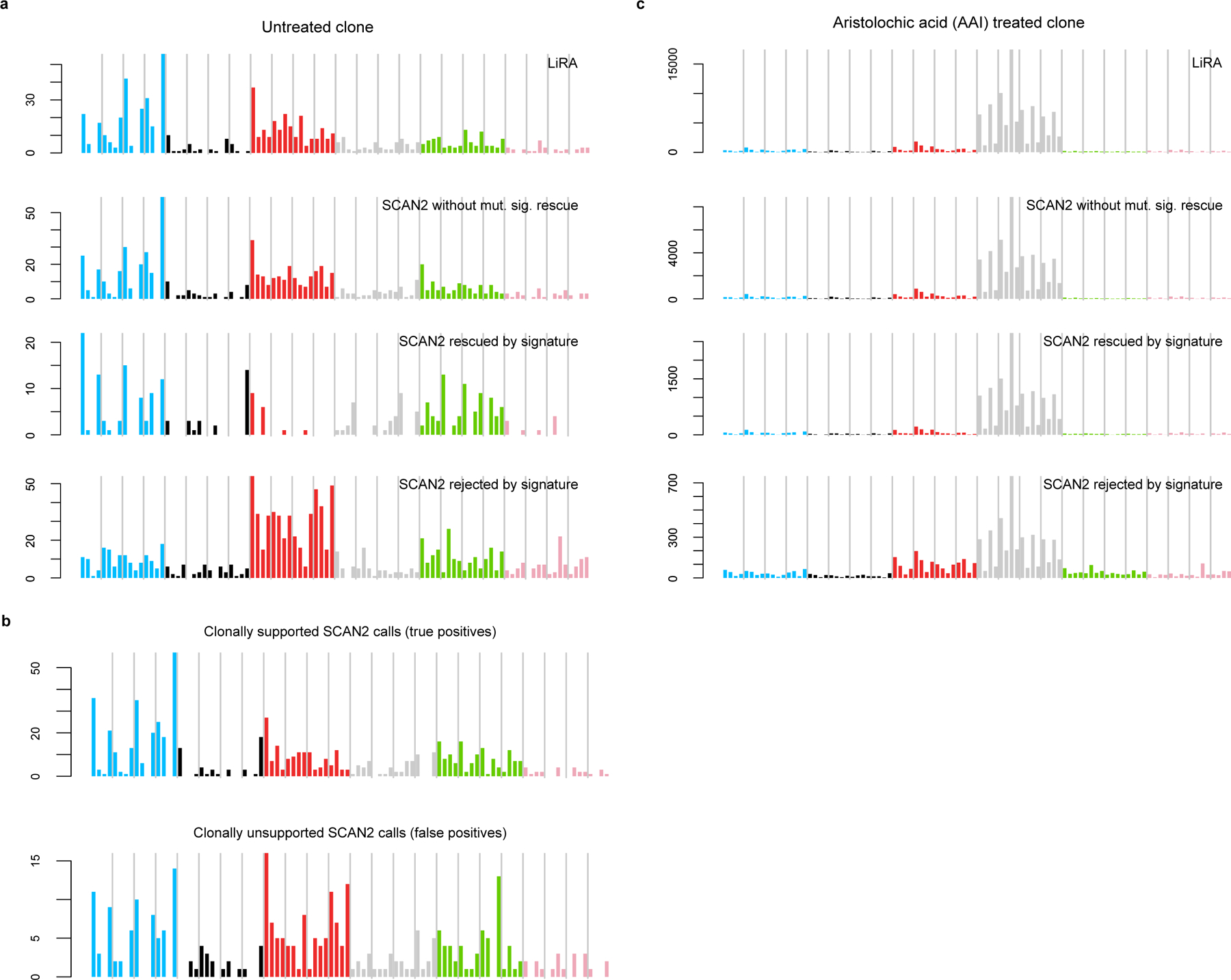
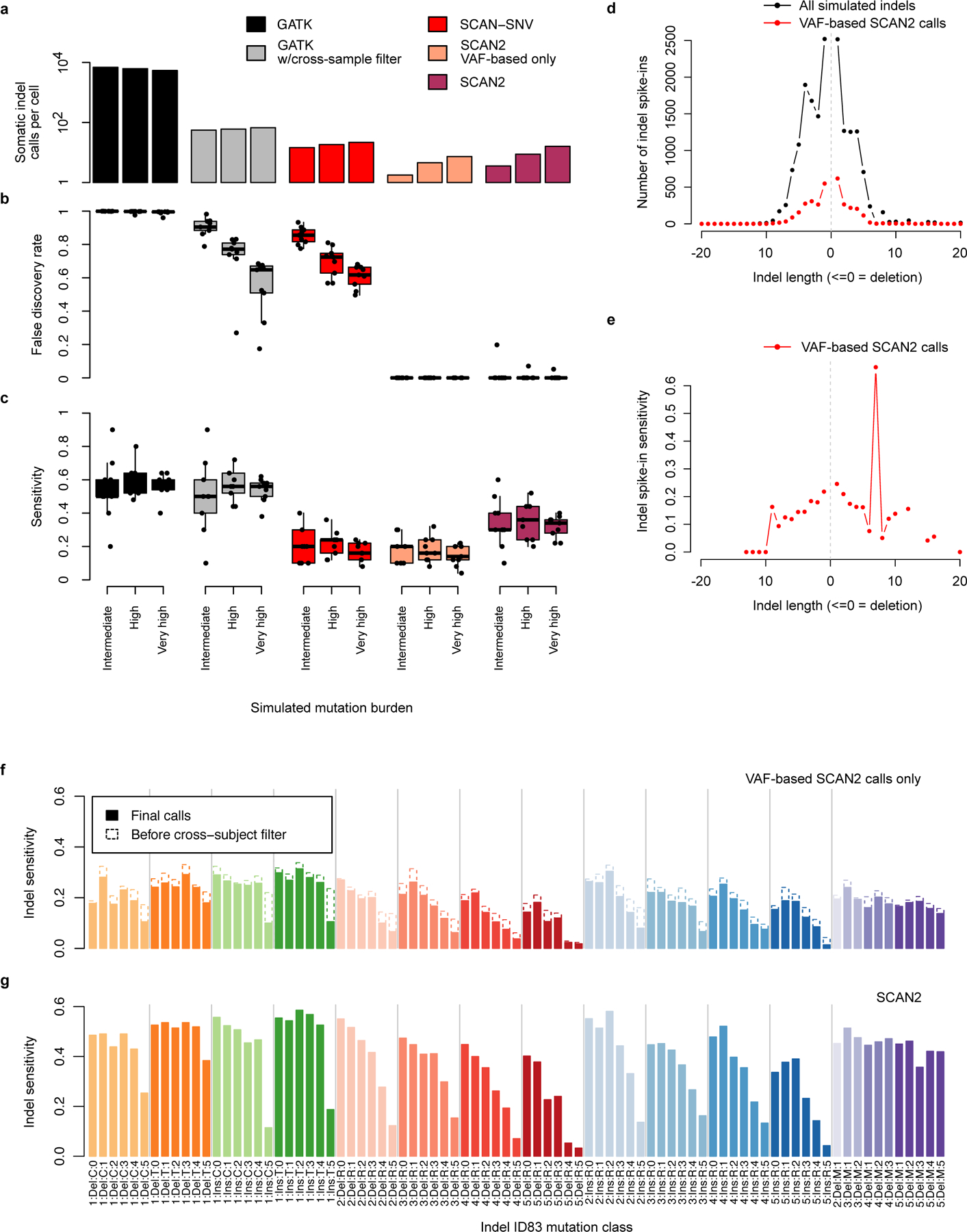
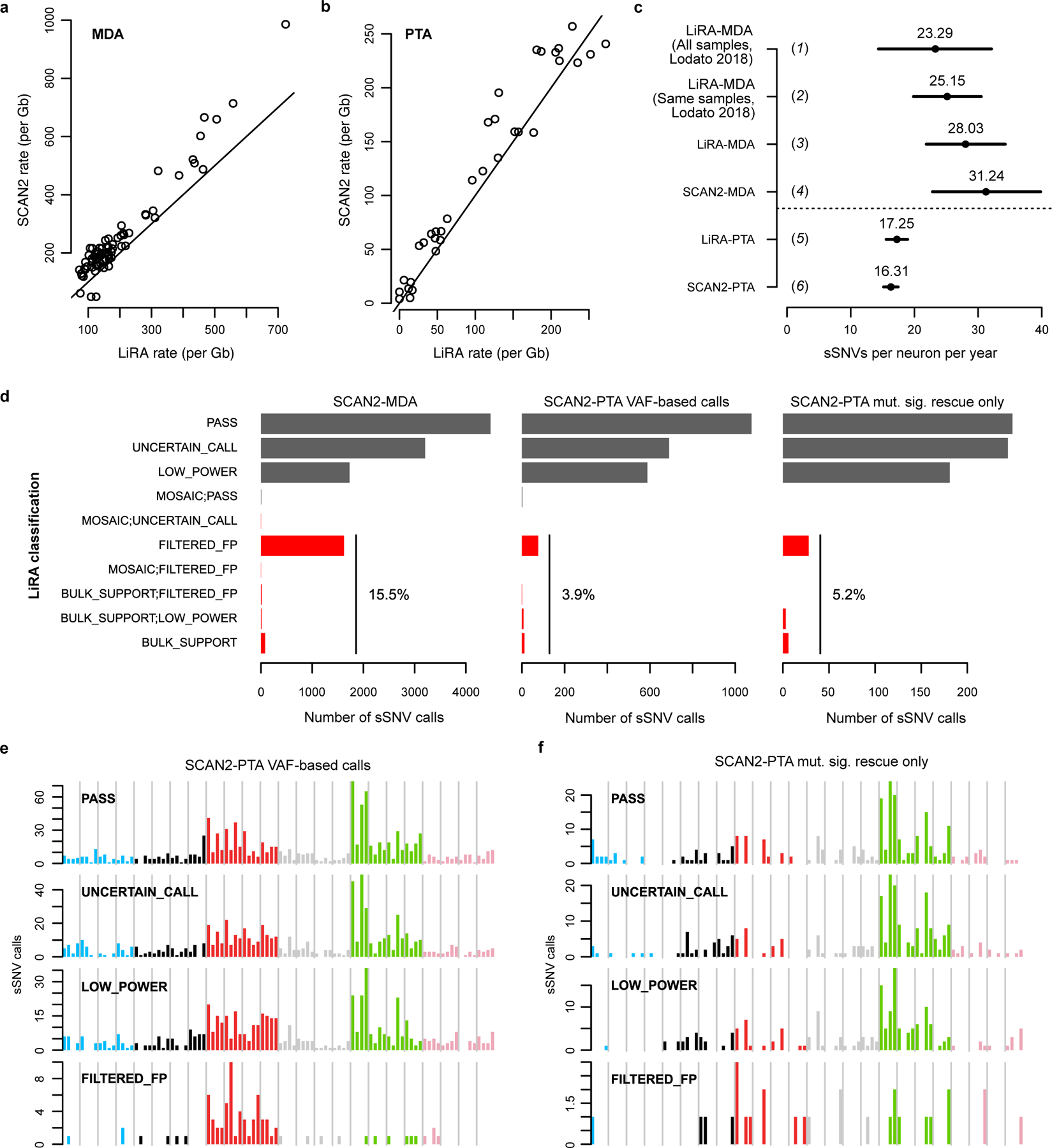
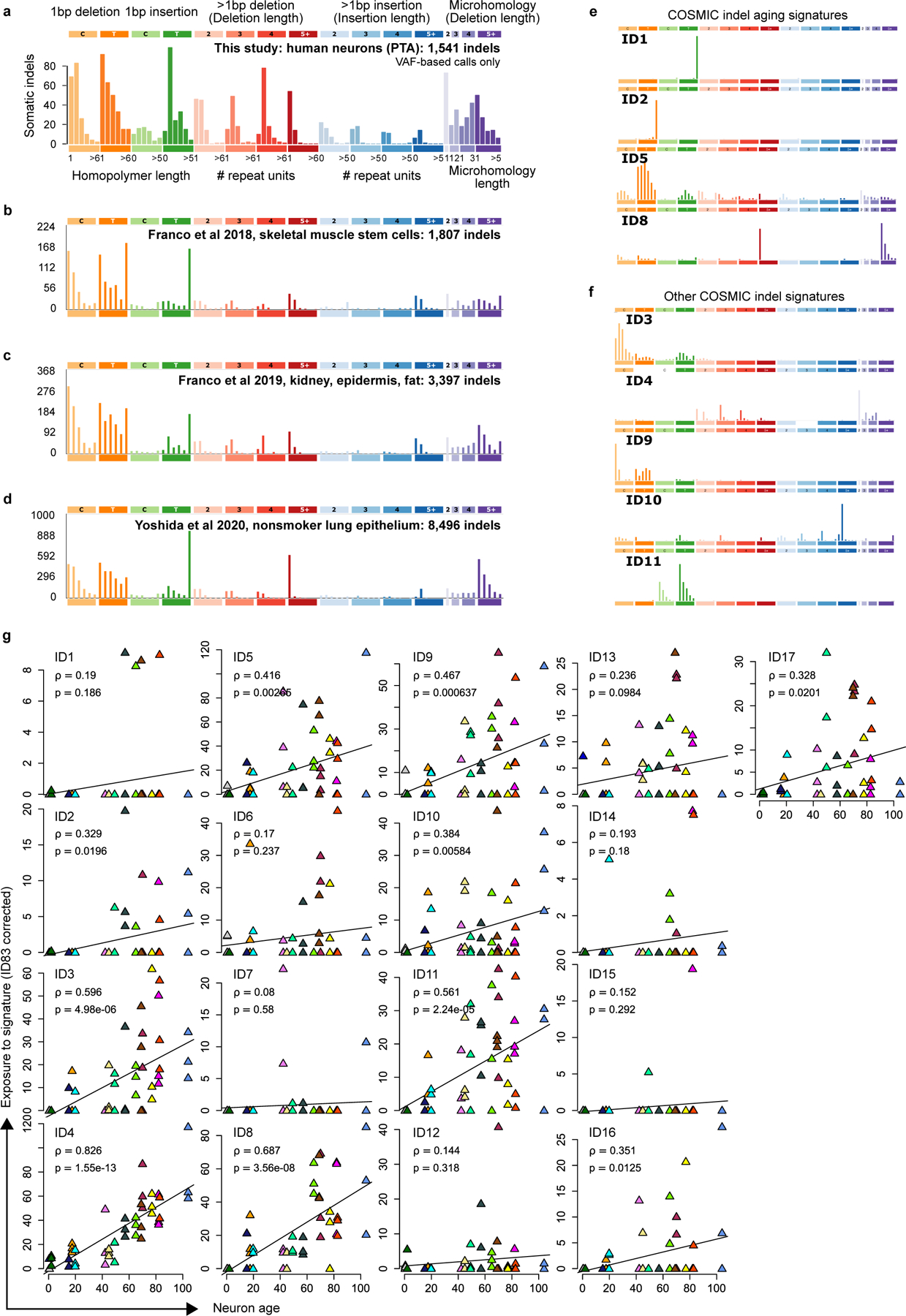
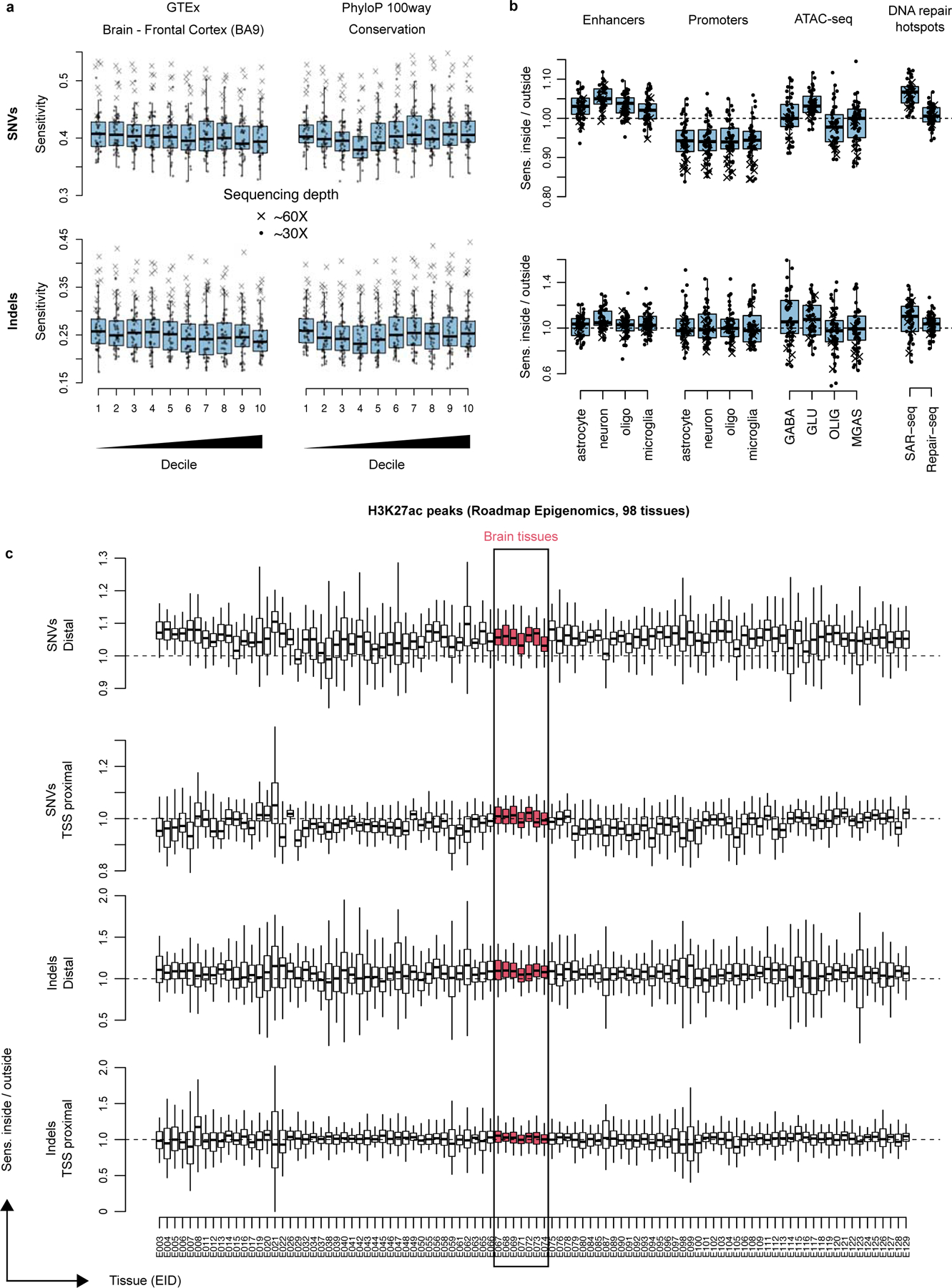
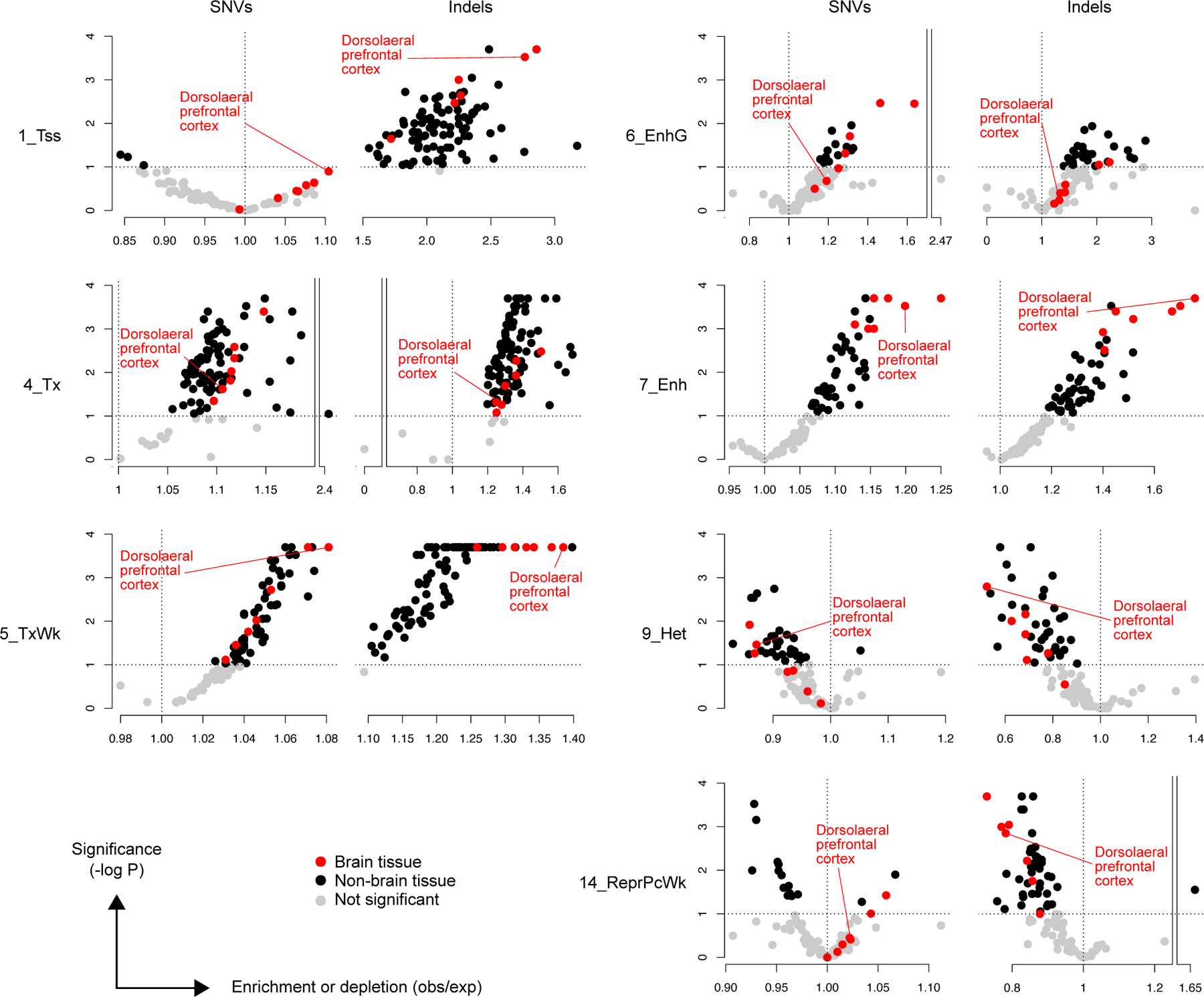
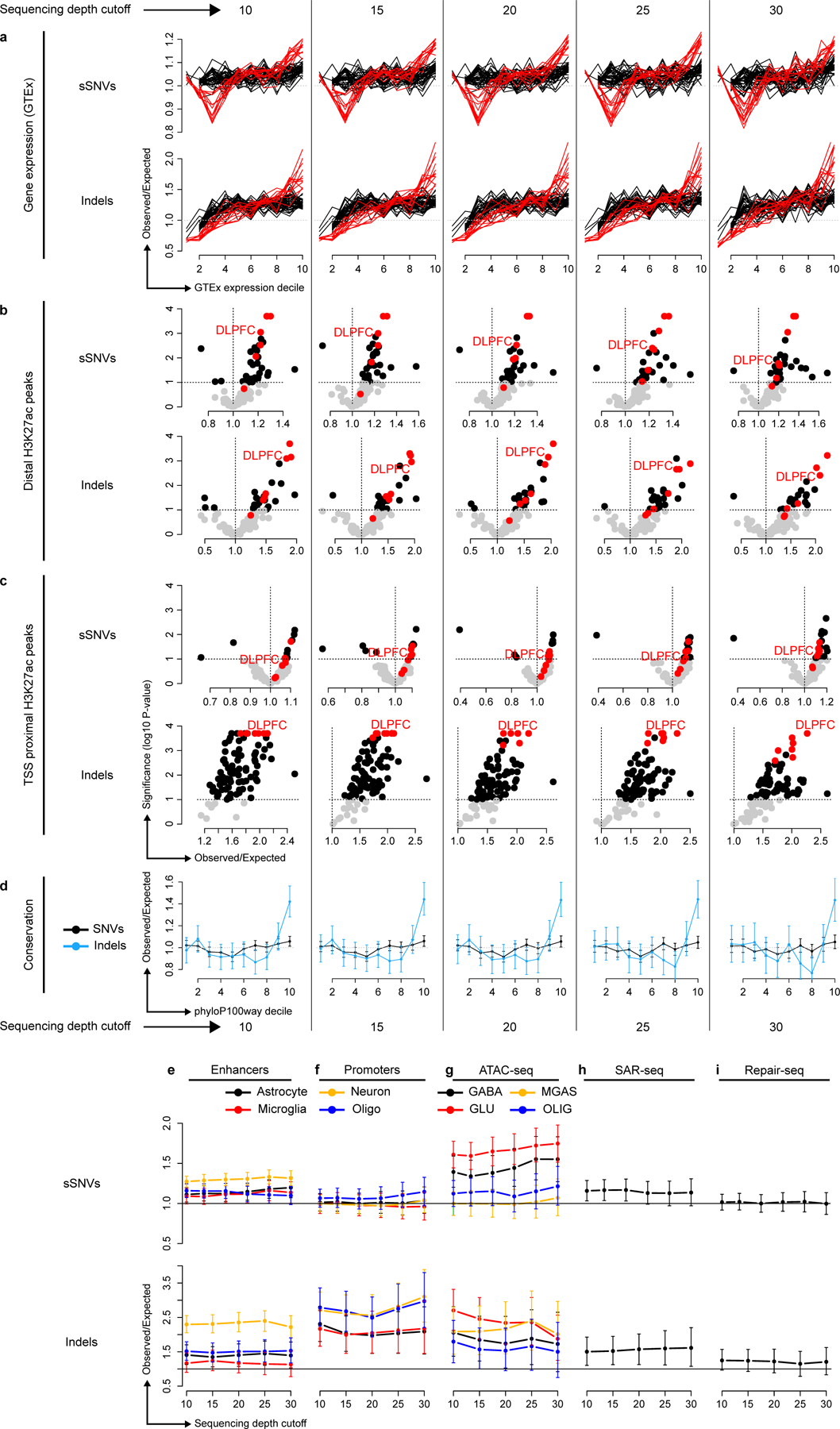
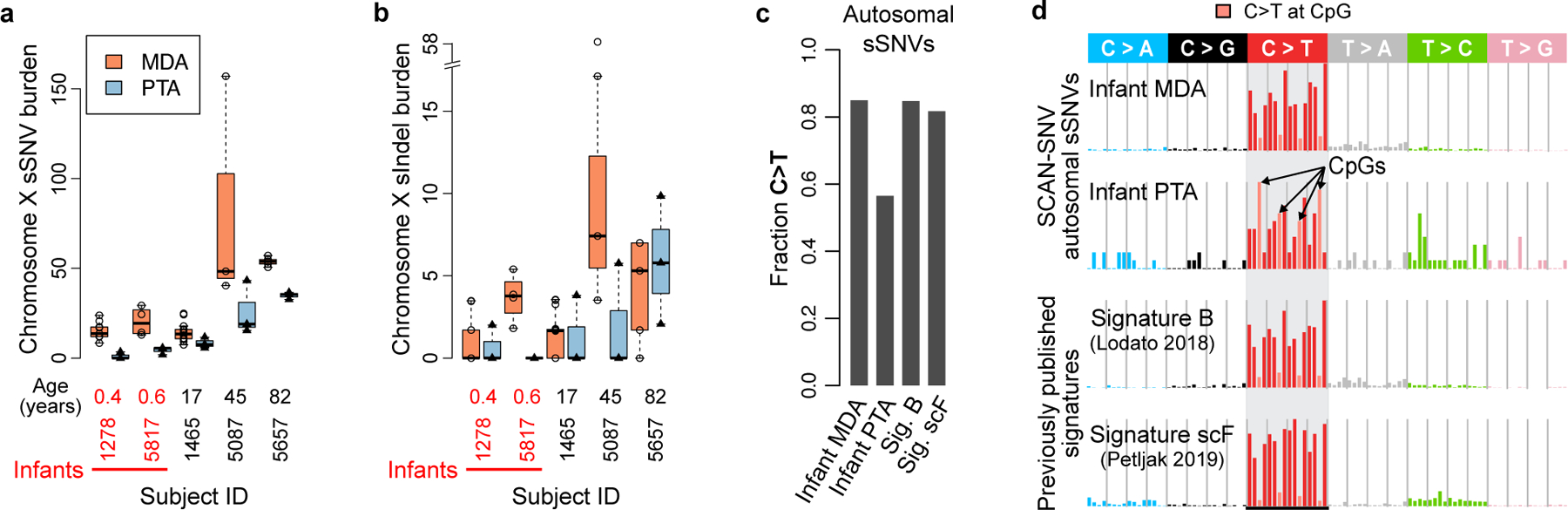
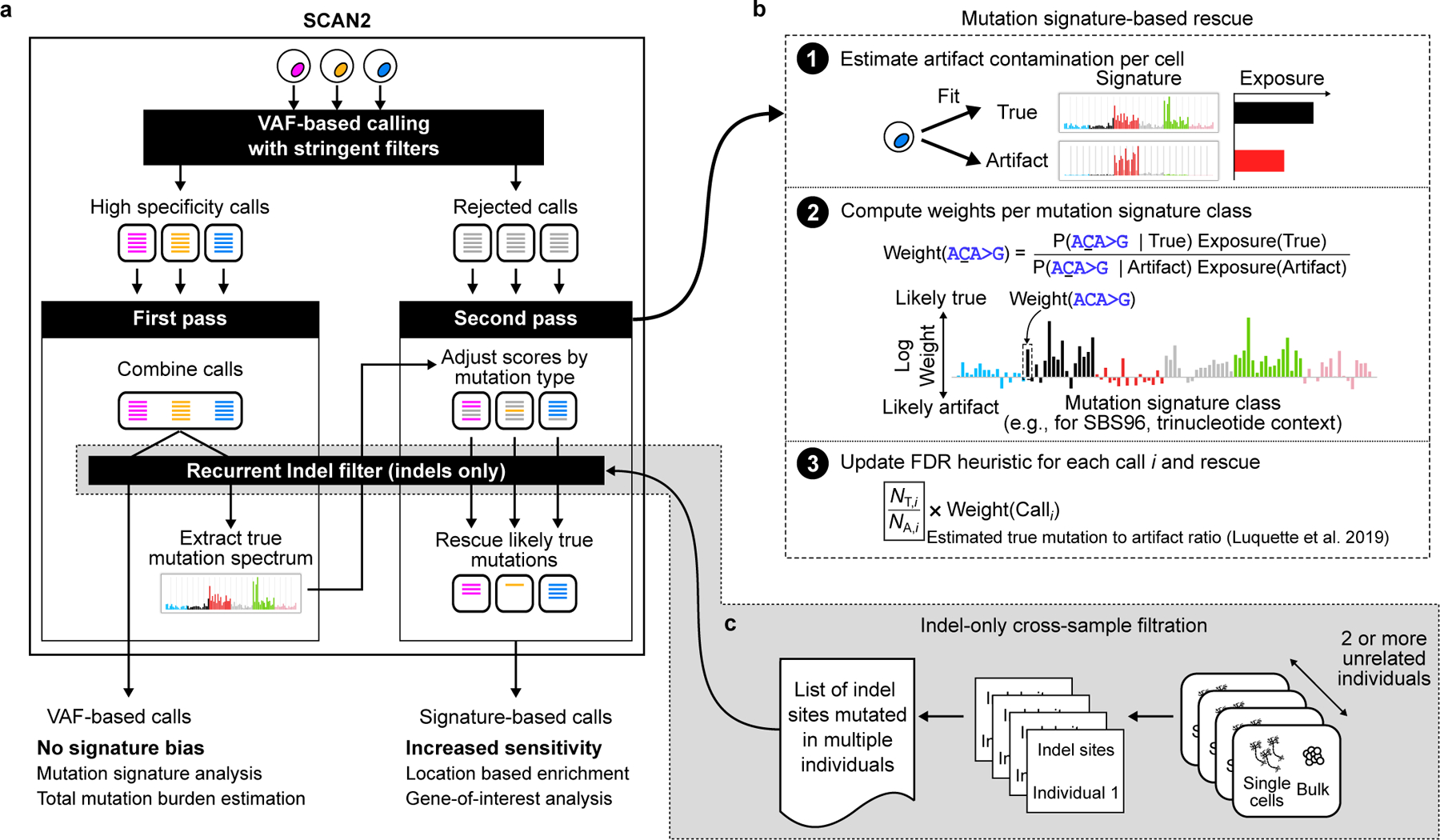
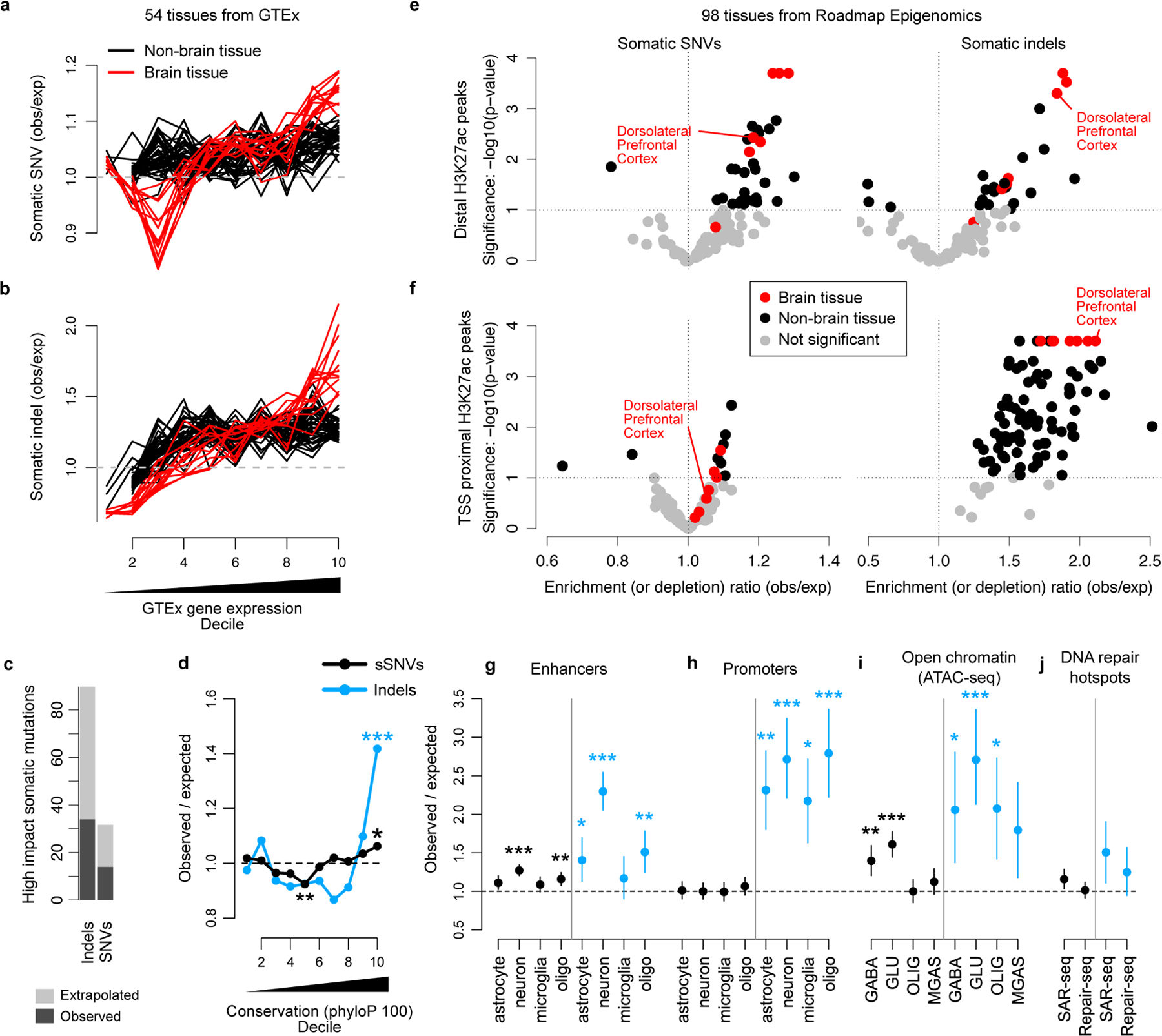
Accurate somatic mutation detection from single-cell DNA sequencing is challenging due to amplification-related artifacts. To reduce this artifact burden, an improved amplification technique, primary template-directed amplification (PTA), was recently introduced. We analyzed whole-genome sequencing data from 52 PTA-amplified single neurons using SCAN2, a new genotyper we developed to leverage mutation signatures and allele balance in identifying somatic single-nucleotide variants (SNVs) and small insertions and deletions (indels) in PTA data. Our analysis confirms an increase in nonclonal somatic mutation in single neurons with age, but revises the estimated rate of this accumulation to 16 SNVs per year. We also identify artifacts in other amplification methods. Most importantly, we show that somatic indels increase by at least three per year per neuron and are enriched in functional regions of the genome such as enhancers and promoters. Our data suggest that indels in gene-regulatory elements have a considerable effect on genome integrity in human neurons.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
