

Discovery and molecular basis of subtype-selective cyclophilin inhibitors
- PMID: 36163383
- PMCID: PMC9596378
- DOI: 10.1038/s41589-022-01116-1
Discovery and molecular basis of subtype-selective cyclophilin inhibitors
Abstract
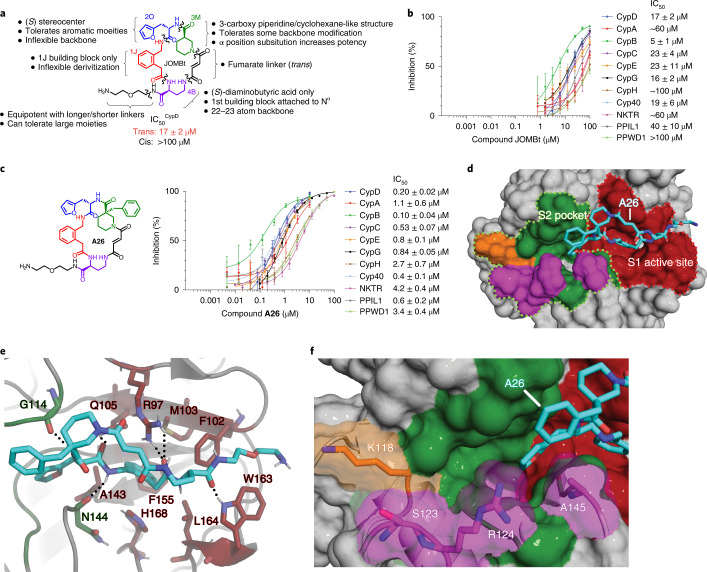
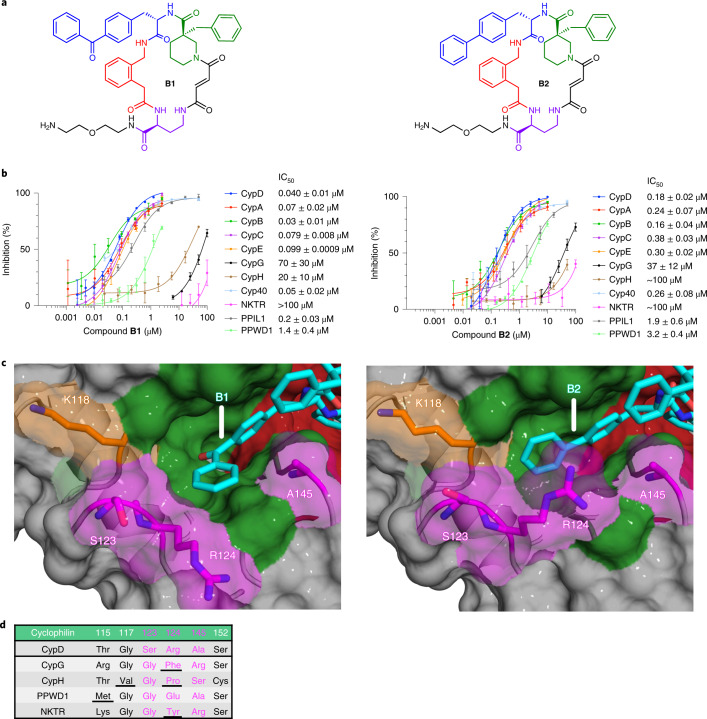
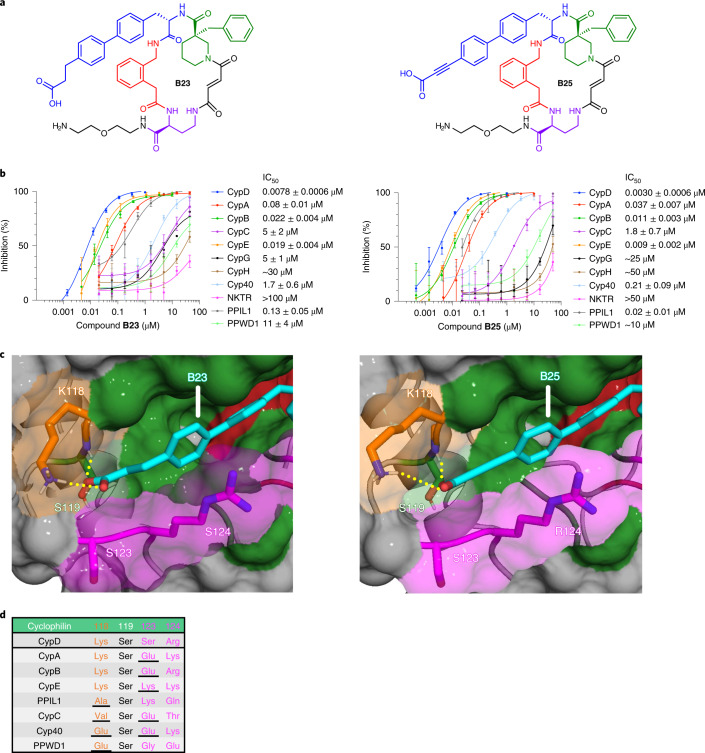
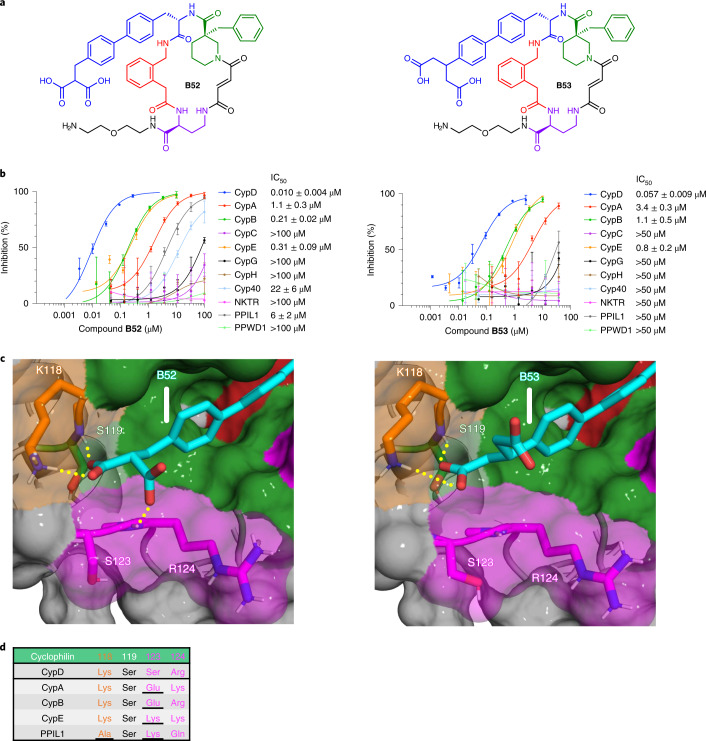
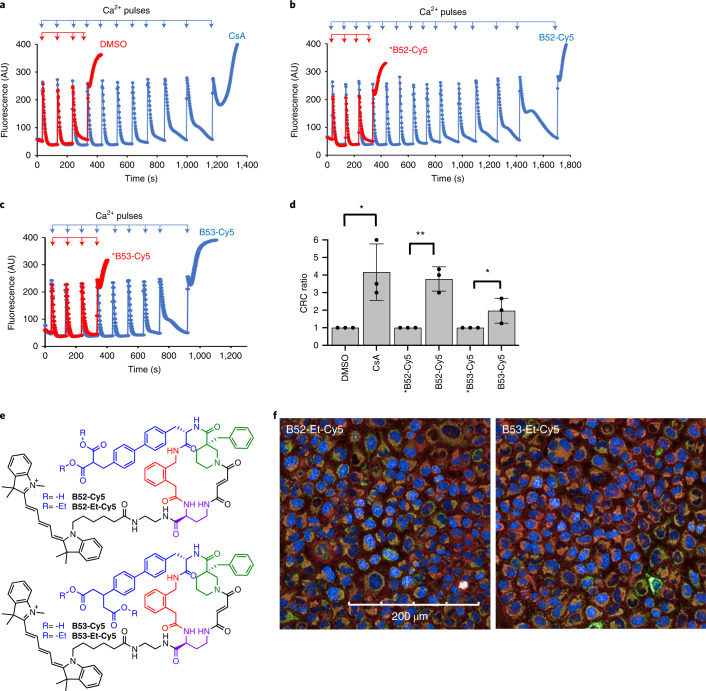
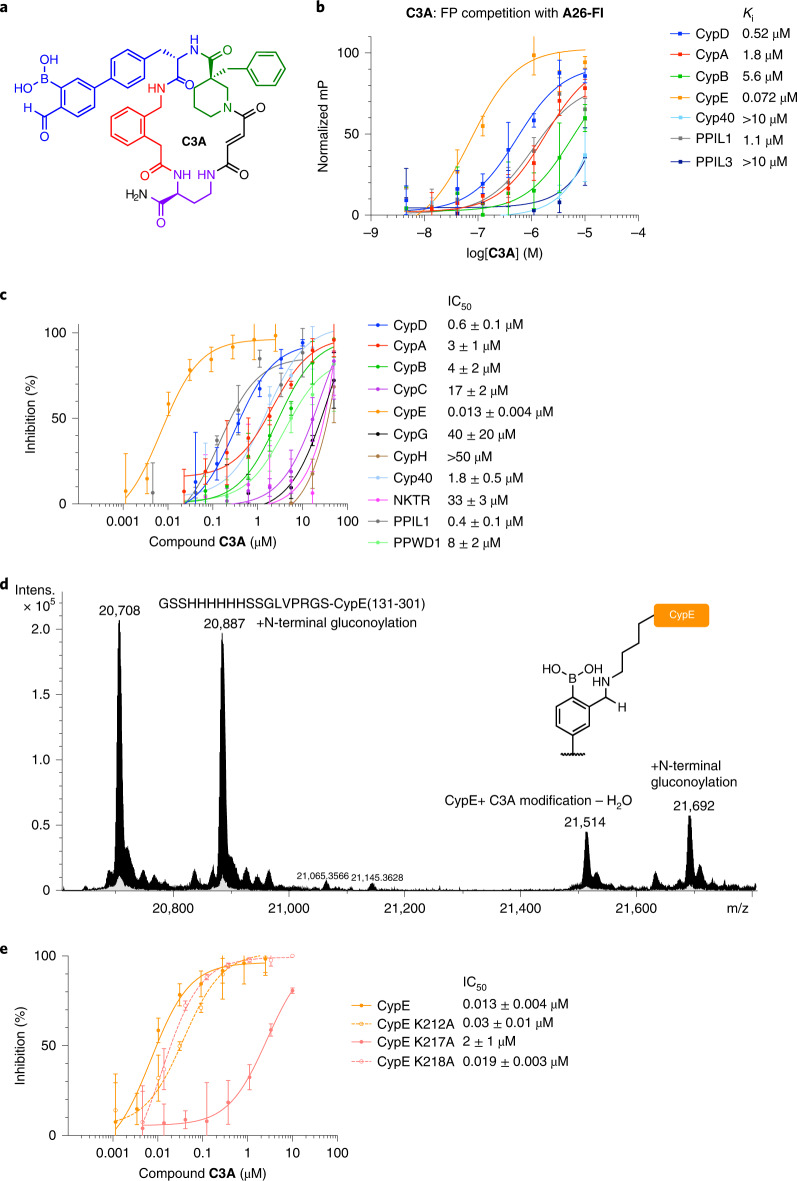
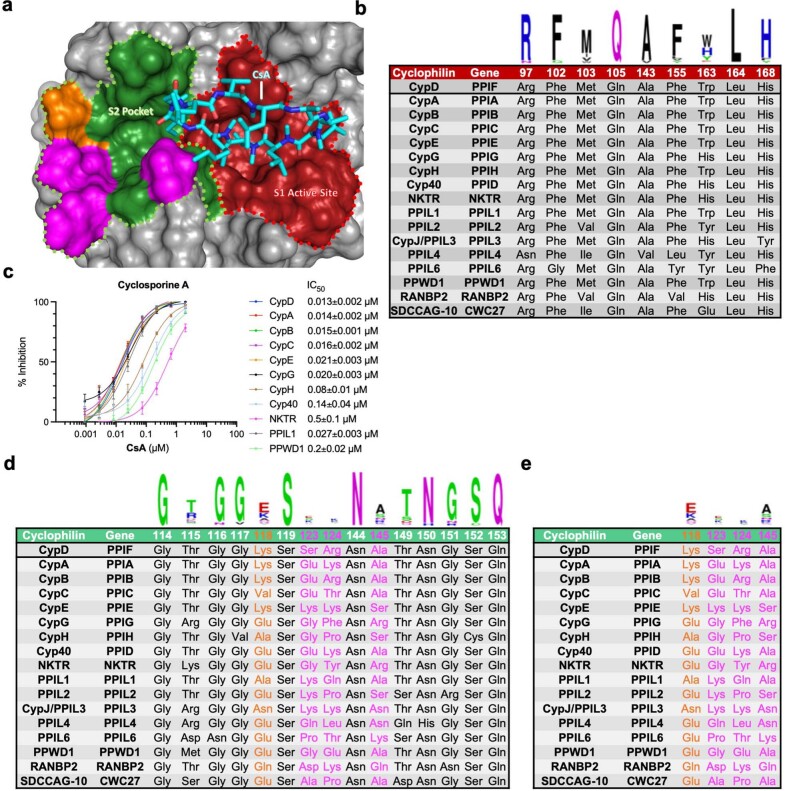
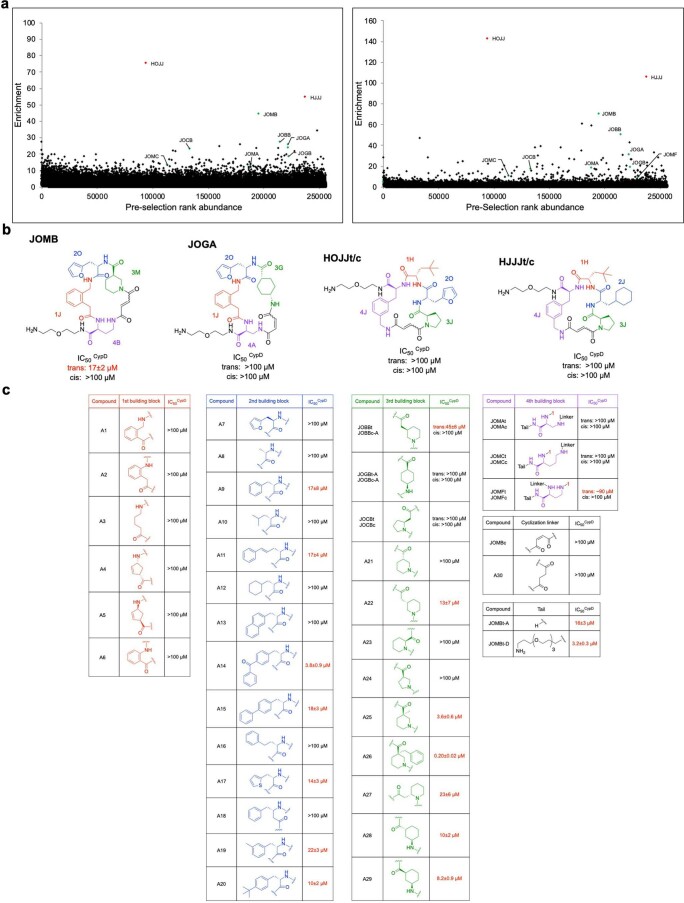
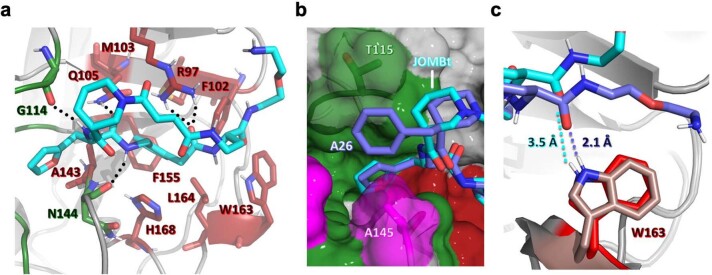
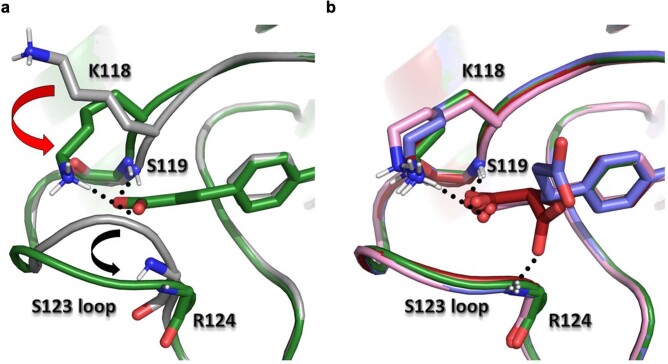
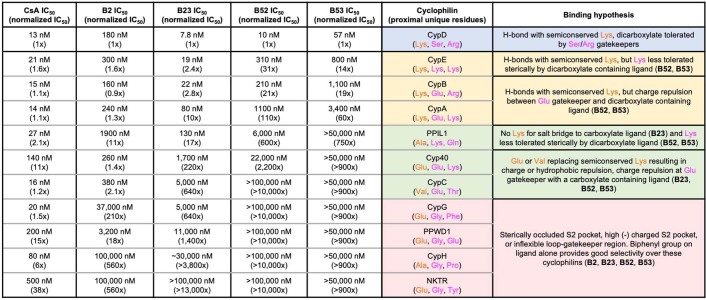
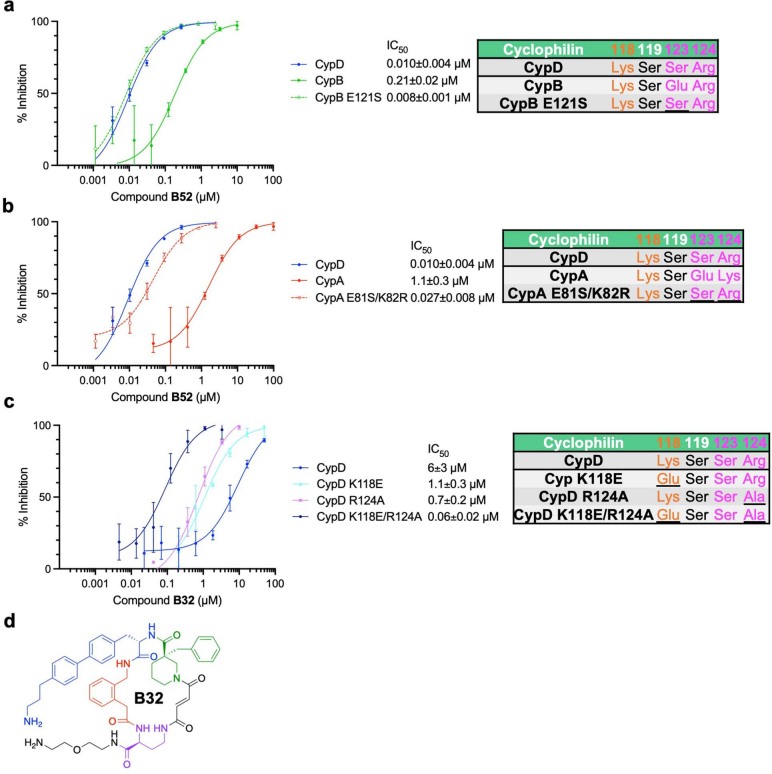
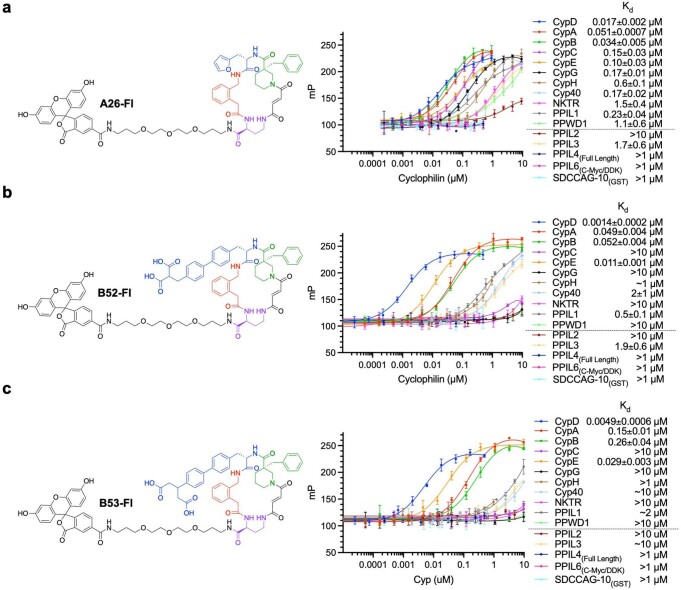
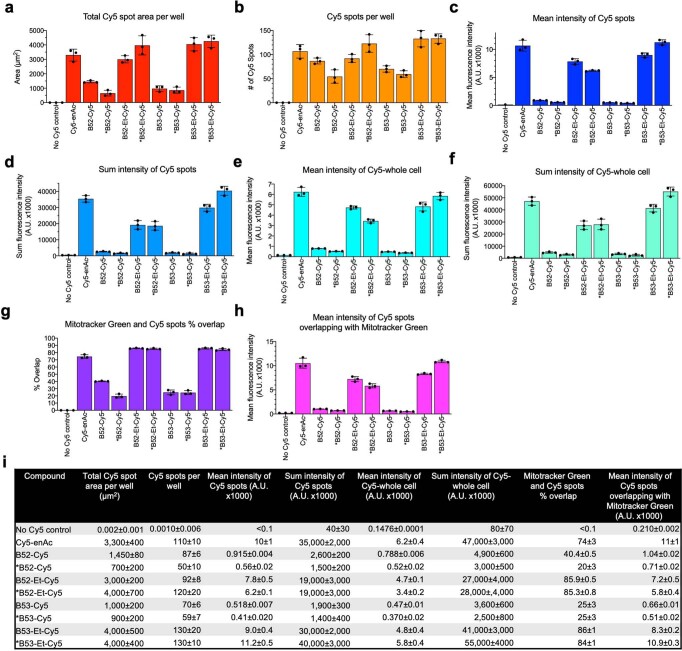
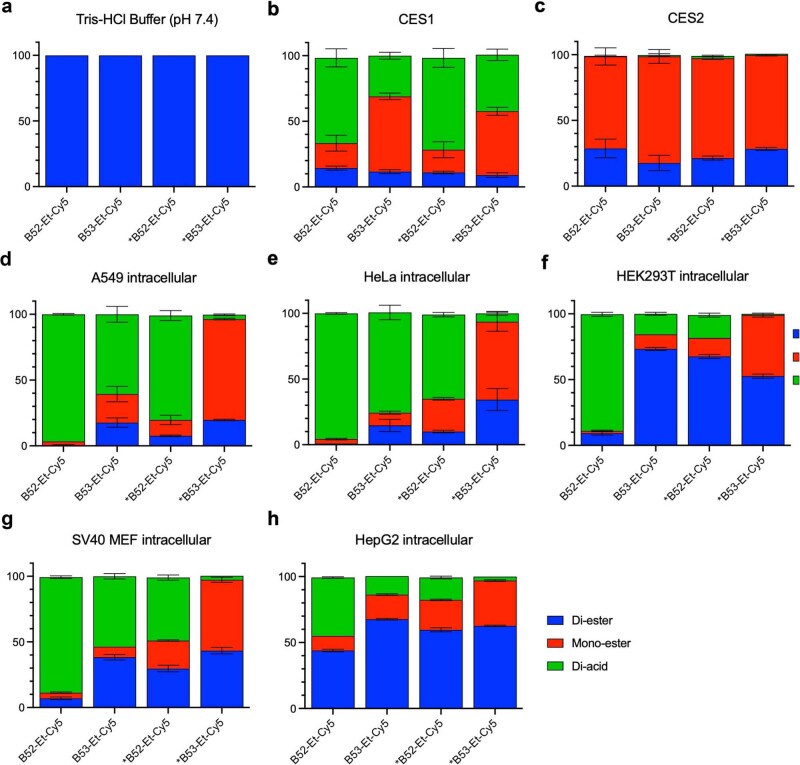
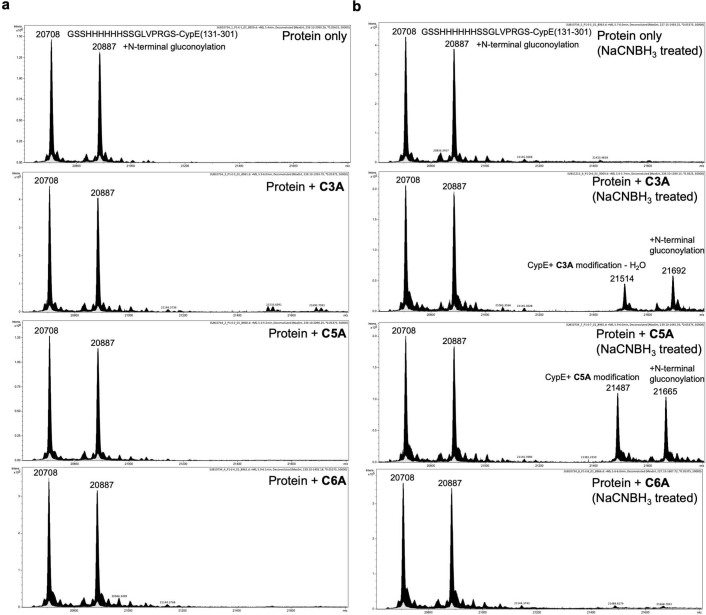
Although cyclophilins are attractive targets for probing biology and therapeutic intervention, no subtype-selective cyclophilin inhibitors have been described. We discovered novel cyclophilin inhibitors from the in vitro selection of a DNA-templated library of 256,000 drug-like macrocycles for cyclophilin D (CypD) affinity. Iterated macrocycle engineering guided by ten X-ray co-crystal structures yielded potent and selective inhibitors (half maximal inhibitory concentration (IC50) = 10 nM) that bind the active site of CypD and also make novel interactions with non-conserved residues in the S2 pocket, an adjacent exo-site. The resulting macrocycles inhibit CypD activity with 21- to >10,000-fold selectivity over other cyclophilins and inhibit mitochondrial permeability transition pore opening in isolated mitochondria. We further exploited S2 pocket interactions to develop the first cyclophilin E (CypE)-selective inhibitor, which forms a reversible covalent bond with a CypE S2 pocket lysine, and exhibits 30- to >4,000-fold selectivity over other cyclophilins. These findings reveal a strategy to generate isoform-selective small-molecule cyclophilin modulators, advancing their suitability as targets for biological investigation and therapeutic development.
© 2022. The Author(s).
Conflict of interest statement
A.A.P. and D.R.L. are co-inventors on patent applications that are based on this work. D.R.L. is a consultant and co-founder of Exo Therapeutics, a company that uses DNA-encoded libraries and exo-site inhibition.
Figures
References
-
- Chen J, et al. Biochemical features of recombinant human cyclophilin J. Anticancer Res. 2016;36:1175–1180. - PubMed
-
- Austin, C. et al. Prolyl Isomerases—Old Proteins as New Therapeutic Targets (Selcia Ltd, 2015).
-
- Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. 2016;26:655–667. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
