

Translational enhancement by base editing of the Kozak sequence rescues haploinsufficiency
- PMID: 36165847
- PMCID: PMC9561285
- DOI: 10.1093/nar/gkac799
Translational enhancement by base editing of the Kozak sequence rescues haploinsufficiency
Abstract
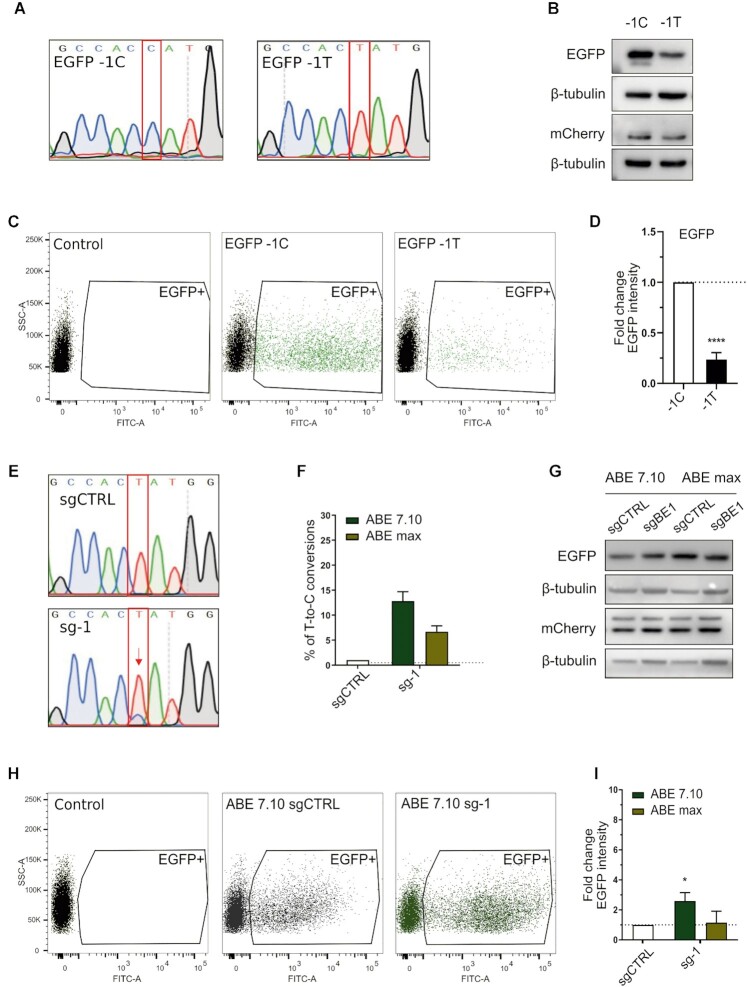
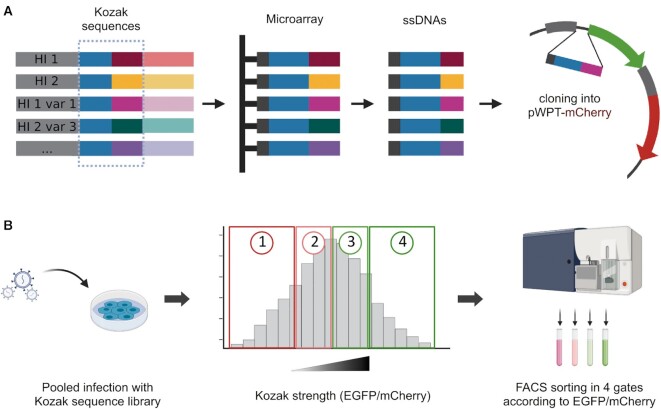
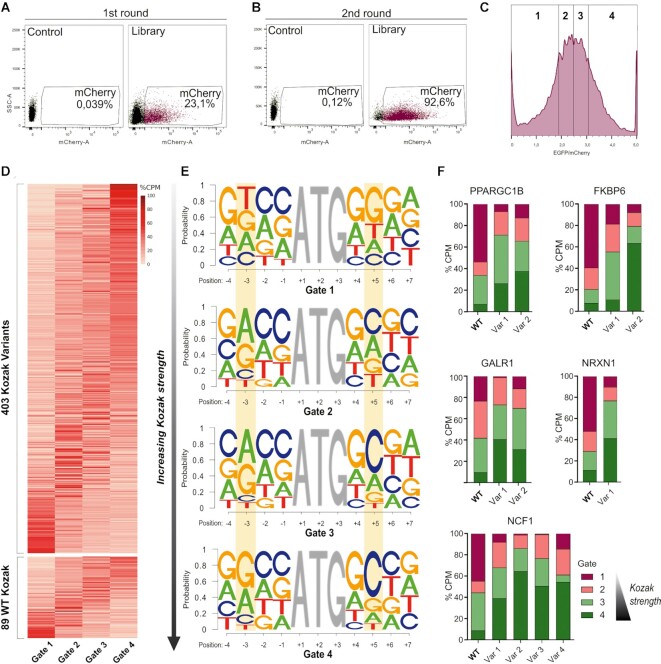
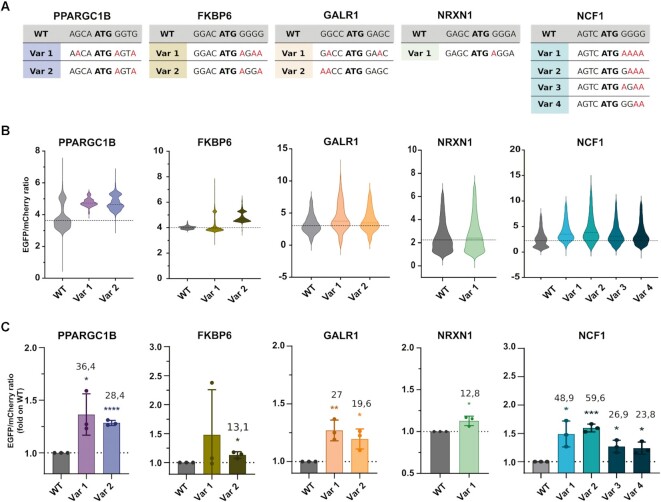
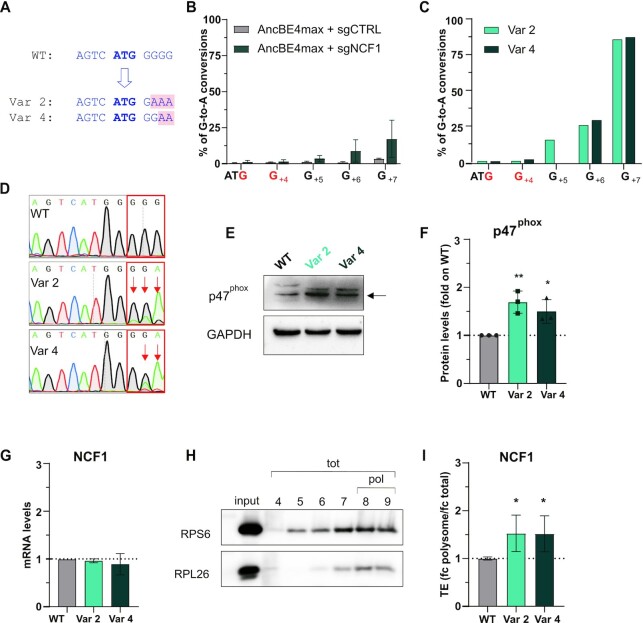
A variety of single-gene human diseases are caused by haploinsufficiency, a genetic condition by which mutational inactivation of one allele leads to reduced protein levels and functional impairment. Translational enhancement of the spare allele could exert a therapeutic effect. Here we developed BOOST, a novel gene-editing approach to rescue haploinsufficiency loci by the change of specific single nucleotides in the Kozak sequence, which controls translation by regulating start codon recognition. We evaluated for translational strength 230 Kozak sequences of annotated human haploinsufficient genes and 4621 derived variants, which can be installed by base editing, by a high-throughput reporter assay. Of these variants, 149 increased the translation of 47 Kozak sequences, demonstrating that a substantial proportion of haploinsufficient genes are controlled by suboptimal Kozak sequences. Validation of 18 variants for 8 genes produced an average enhancement in an expression window compatible with the rescue of the genetic imbalance. Base editing of the NCF1 gene, whose monoallelic loss causes chronic granulomatous disease, resulted in the desired increase of NCF1 (p47phox) protein levels in a relevant cell model. We propose BOOST as a fine-tuned approach to modulate translation, applicable to the correction of dozens of haploinsufficient monogenic disorders independently of the causing mutation.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Torgerson T., Ochs H.. Genetics of primary immune deficiencies. Stiehm's Immune Defic. 2014; 2014:73–81.
-
- Dang V.T., Kassahn K.S., Marcos A.E., Ragan M.A.. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur. J. Hum. Genet. 2008; 16:1350–1357. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
