

Mutations in protein kinase Cγ promote spinocerebellar ataxia type 14 by impairing kinase autoinhibition
- PMID: 36166510
- PMCID: PMC9810342
- DOI: 10.1126/scisignal.abk1147
Mutations in protein kinase Cγ promote spinocerebellar ataxia type 14 by impairing kinase autoinhibition
Abstract
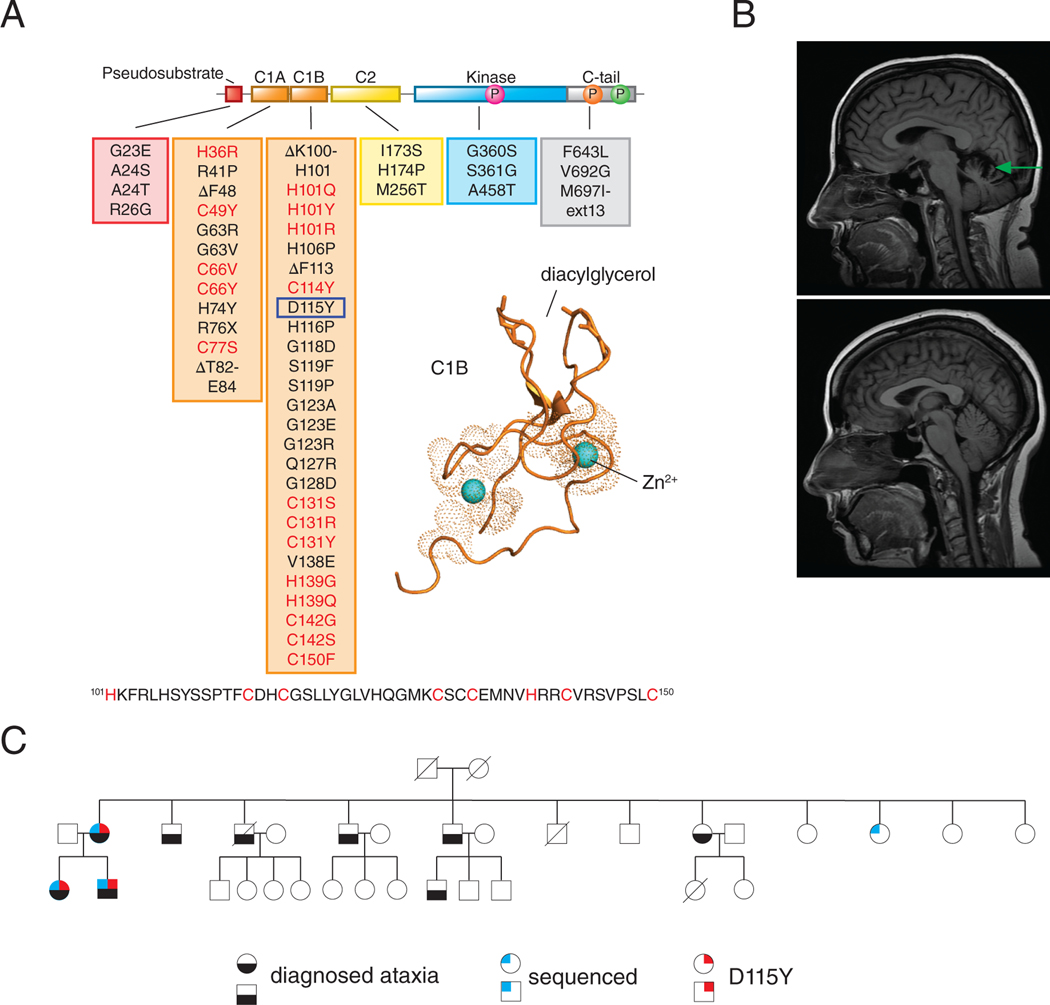
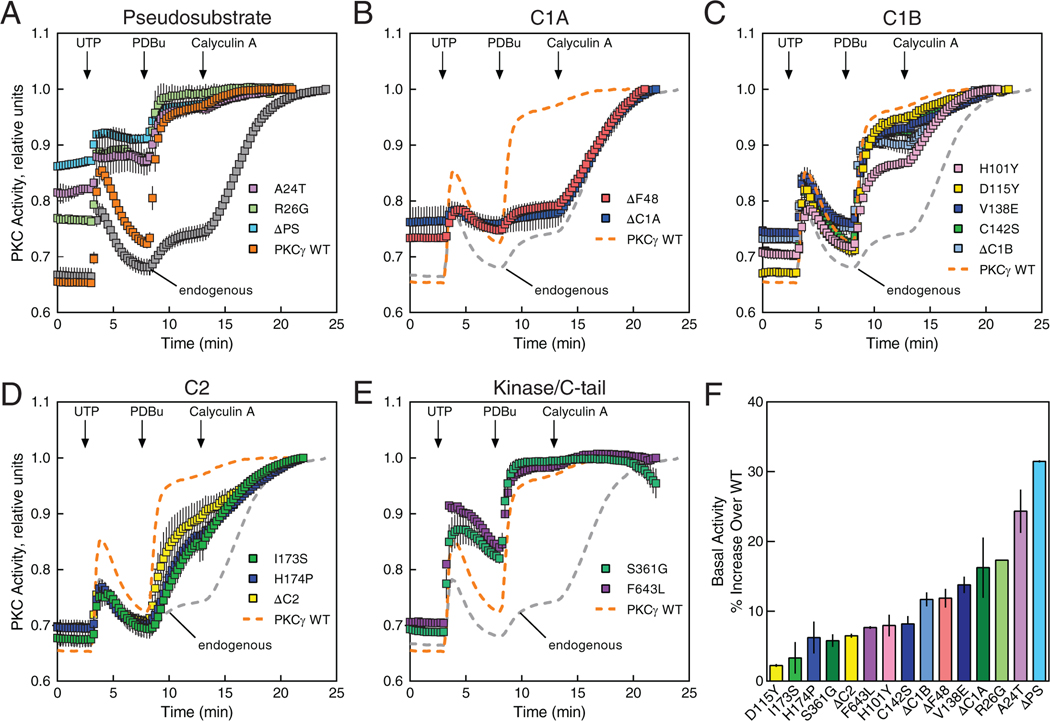
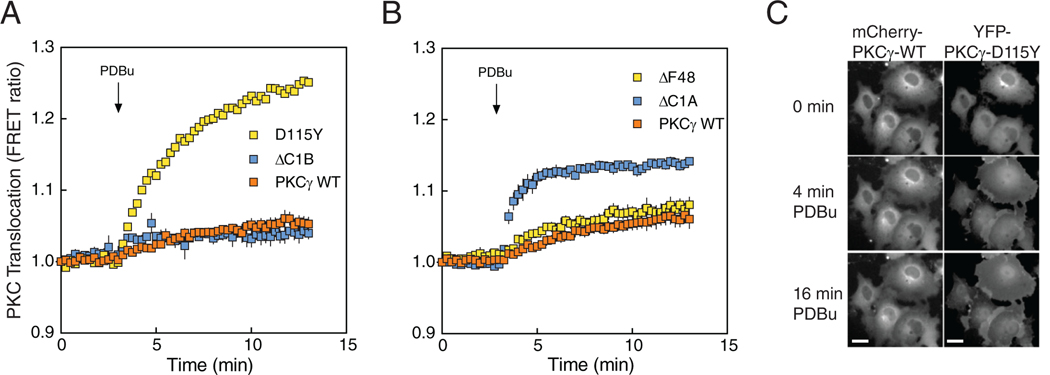
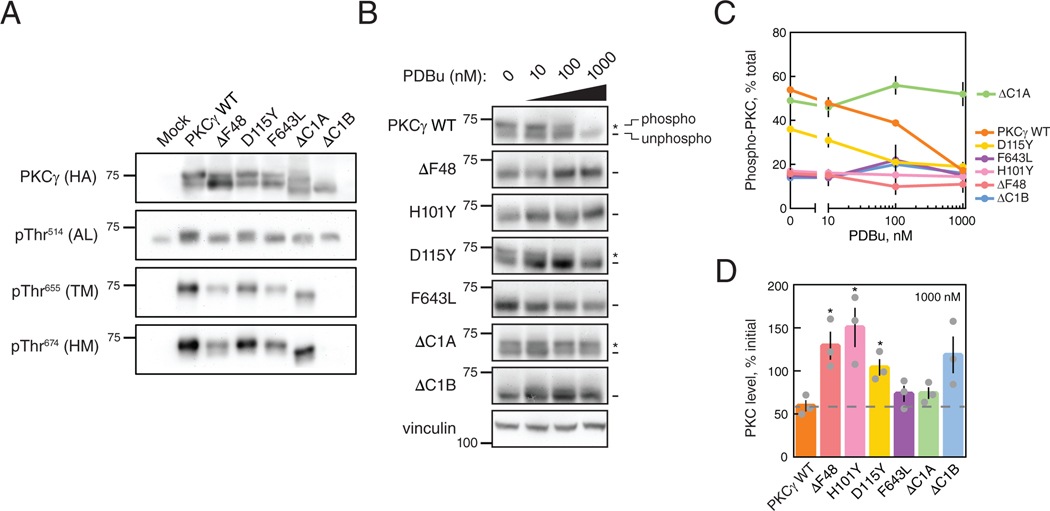
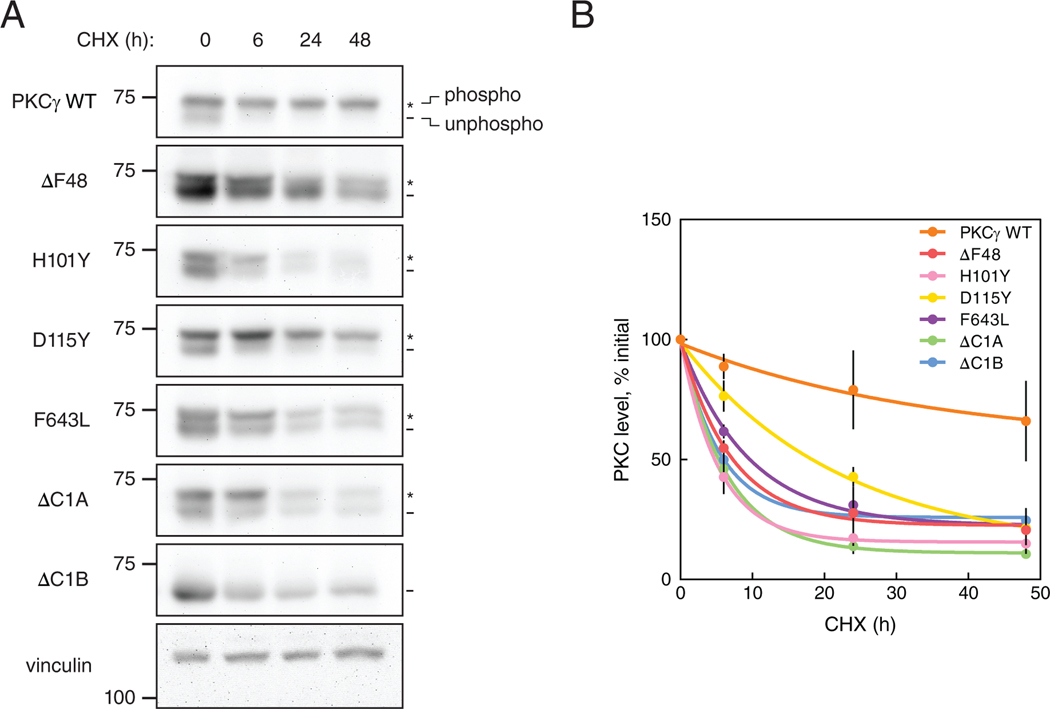
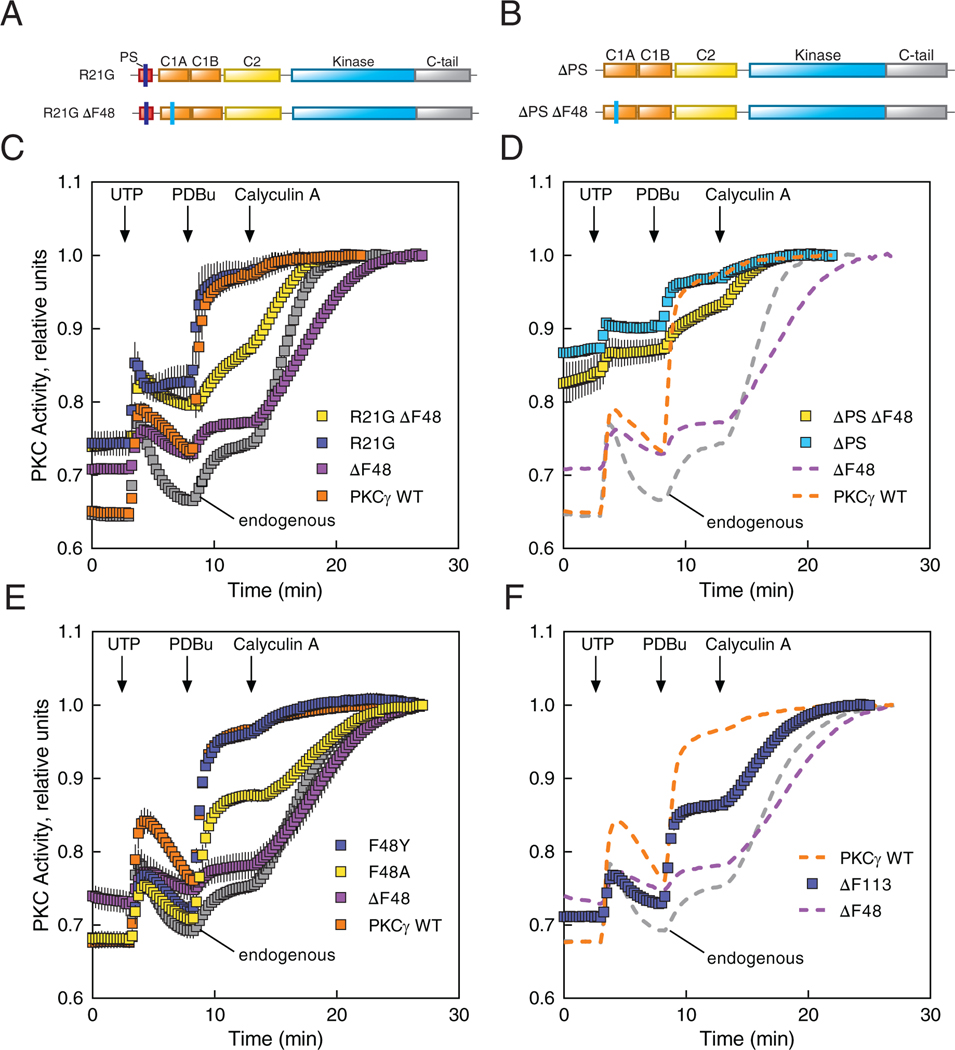
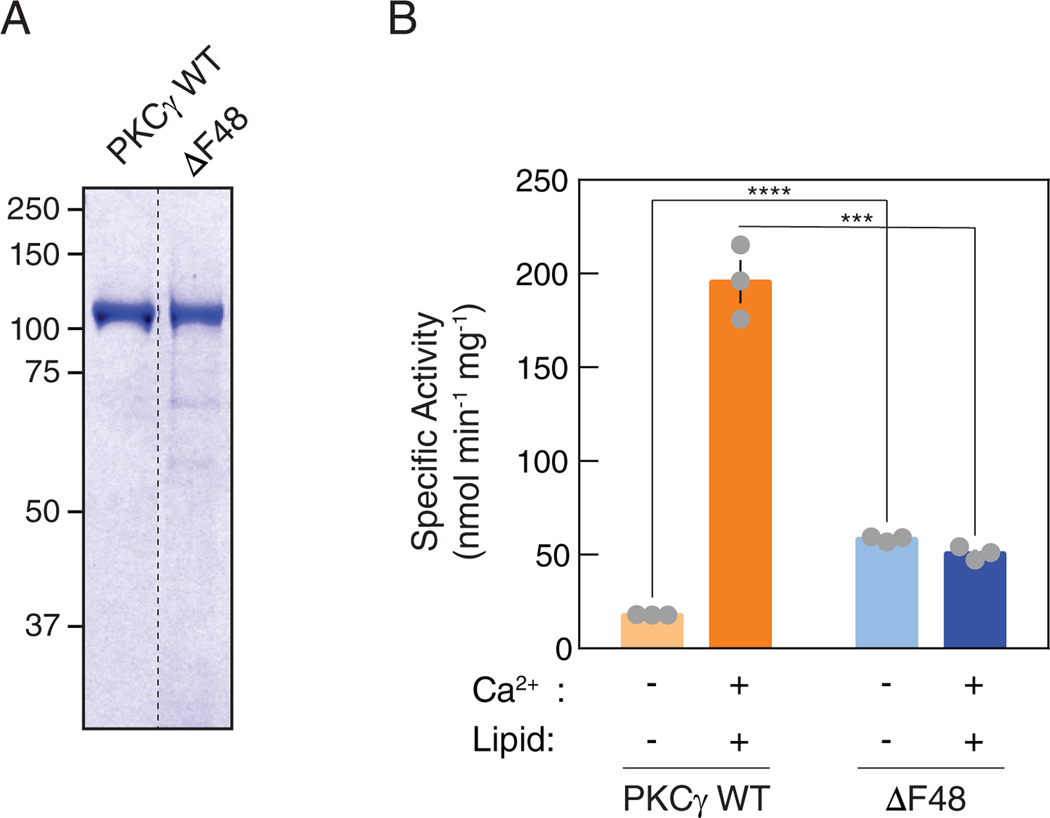
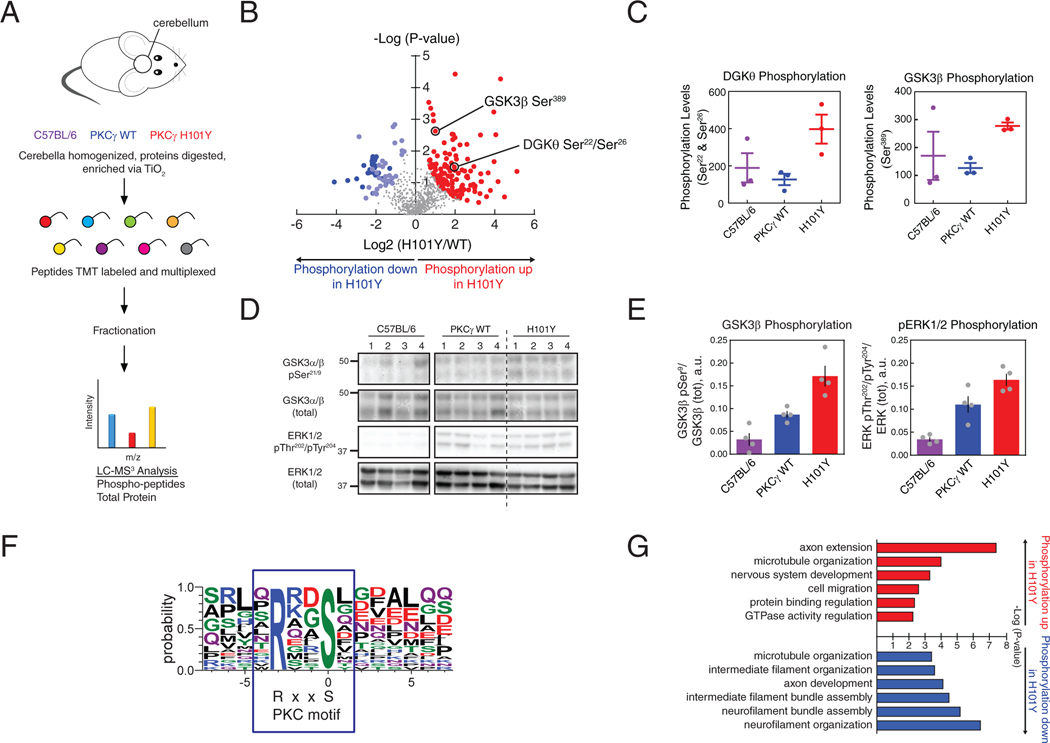
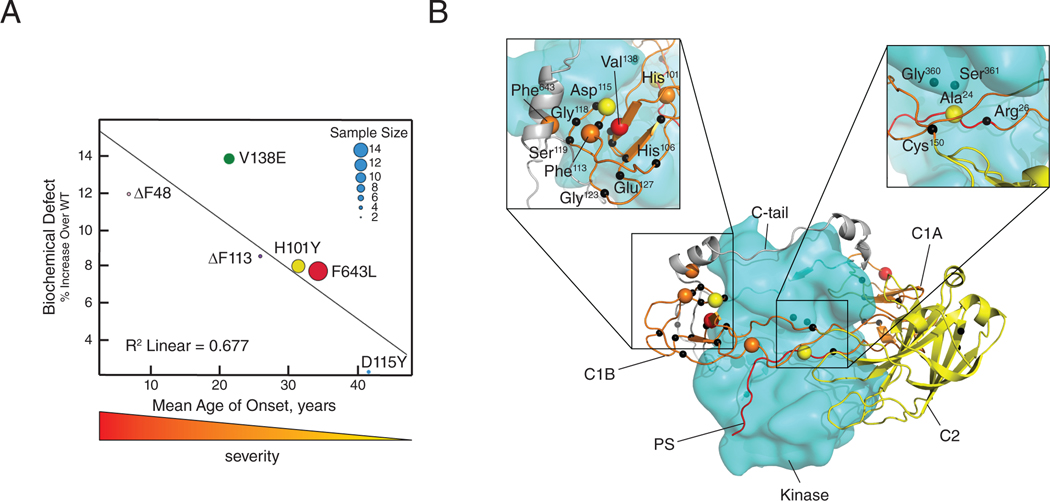
Spinocerebellar ataxia type 14 (SCA14) is a neurodegenerative disease caused by germline variants in the diacylglycerol (DAG)/Ca2+-regulated protein kinase Cγ (PKCγ), leading to Purkinje cell degeneration and progressive cerebellar dysfunction. Most of the identified mutations cluster in the DAG-sensing C1 domains. Here, we found with a FRET-based activity reporter that SCA14-associated PKCγ mutations, including a previously undescribed variant, D115Y, enhanced the basal activity of the kinase by compromising its autoinhibition. Unlike other mutations in PKC that impair its autoinhibition but lead to its degradation, the C1 domain mutations protected PKCγ from such down-regulation. This enhanced basal signaling rewired the brain phosphoproteome, as revealed by phosphoproteomic analysis of cerebella from mice expressing a human SCA14-associated H101Y mutant PKCγ transgene. Mutations that induced a high basal activity in vitro were associated with earlier average age of onset in patients. Furthermore, the extent of disrupted autoinhibition, but not agonist-stimulated activity, correlated with disease severity. Molecular modeling indicated that almost all SCA14 variants not within the C1 domain were located at interfaces with the C1B domain, suggesting that mutations in and proximal to the C1B domain are a susceptibility for SCA14 because they uniquely enhance PKCγ basal activity while protecting the enzyme from down-regulation. These results provide insight into how PKCγ activation is modulated and how deregulation of the cerebellar phosphoproteome by SCA14-associated mutations affects disease progression.
Conflict of interest statement
Figures
References
-
- Nishizuka Y, Protein kinase C and lipid signaling for sustained cellular responses. The FASEB Journal. 9, 484–496 (1995). - PubMed
-
- Callender JA, Yang Y, Lordén G, Stephenson NL, Jones AC, Brognard J, Newton AC, Protein kinase Cα gain-of-function variant in Alzheimer’s disease displays enhanced catalysis by a mechanism that evades down-regulation. Proceedings of the National Academy of Sciences of the United States of America. 115, E5497–E5505 (2018). - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
