

False gene and chromosome losses in genome assemblies caused by GC content variation and repeats
- PMID: 36167554
- PMCID: PMC9516821
- DOI: 10.1186/s13059-022-02765-0
False gene and chromosome losses in genome assemblies caused by GC content variation and repeats
Abstract
Background: Many short-read genome assemblies have been found to be incomplete and contain mis-assemblies. The Vertebrate Genomes Project has been producing new reference genome assemblies with an emphasis on being as complete and error-free as possible, which requires utilizing long reads, long-range scaffolding data, new assembly algorithms, and manual curation. A more thorough evaluation of the recent references relative to prior assemblies can provide a detailed overview of the types and magnitude of improvements.
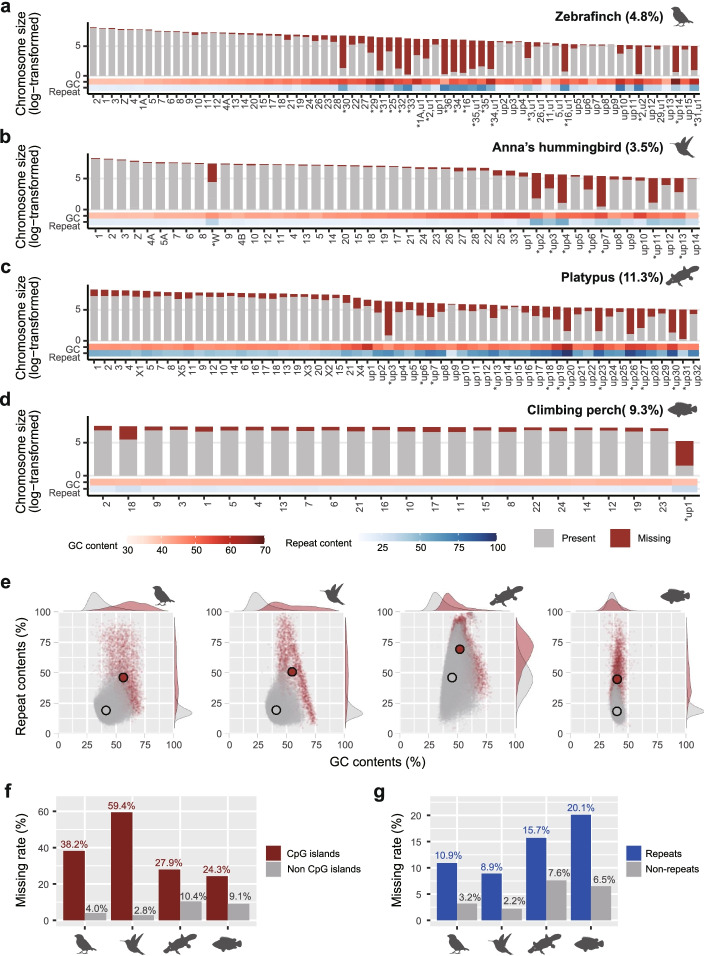
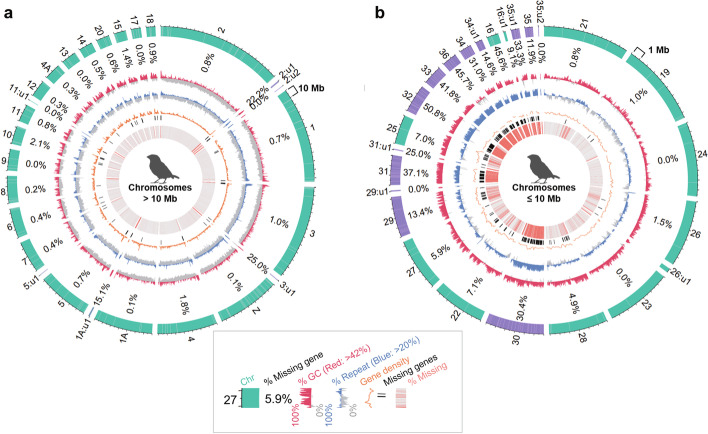
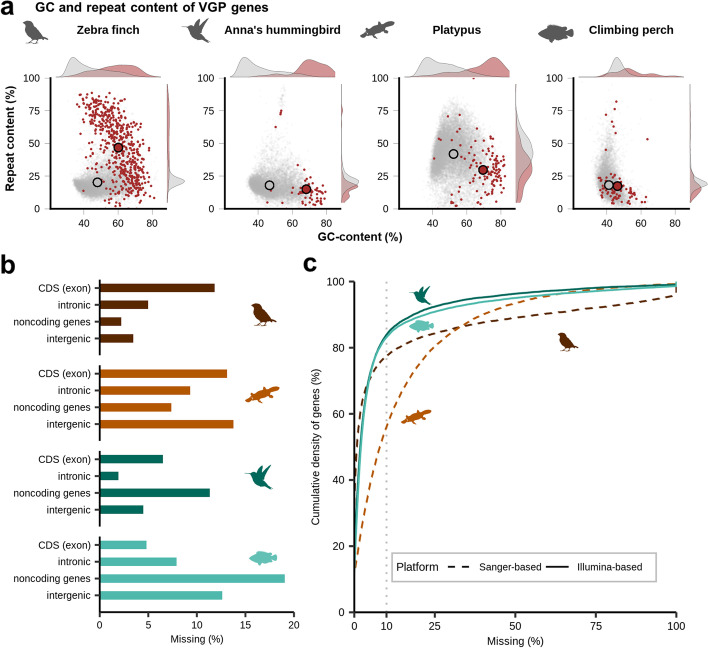
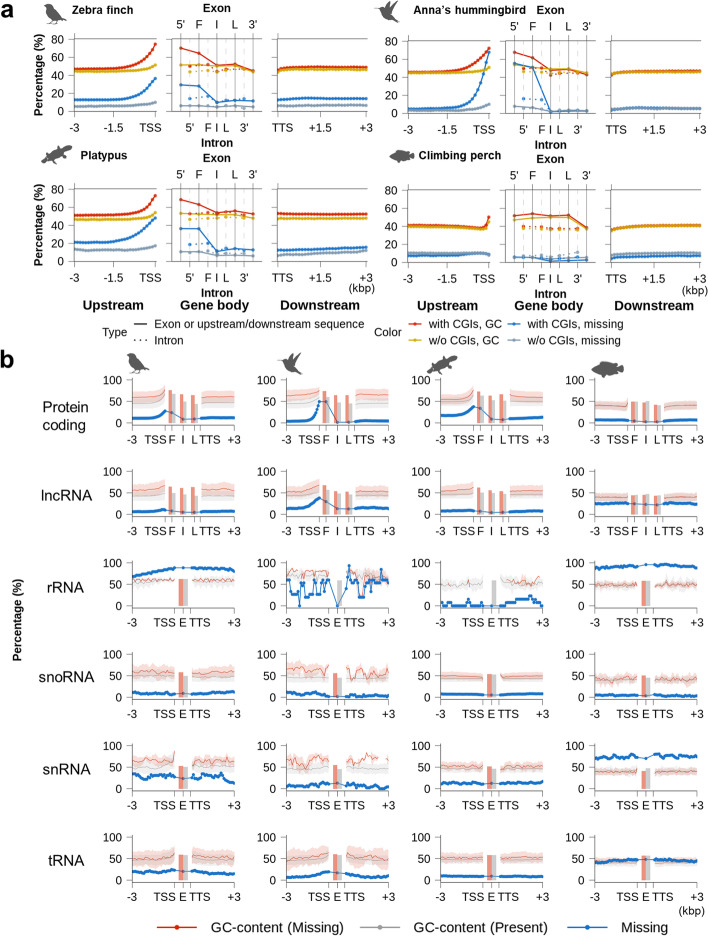
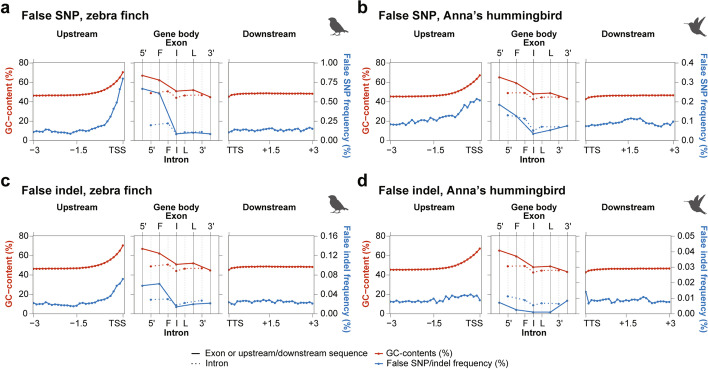
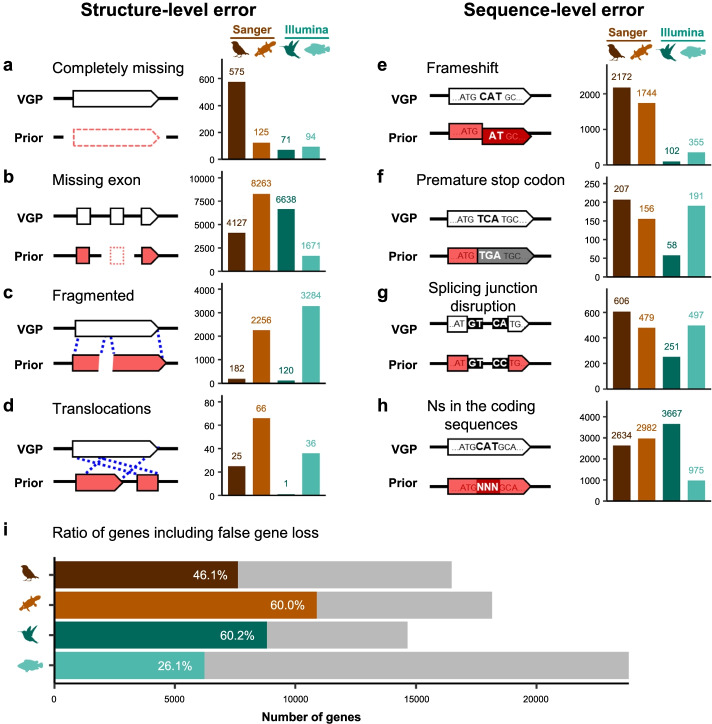
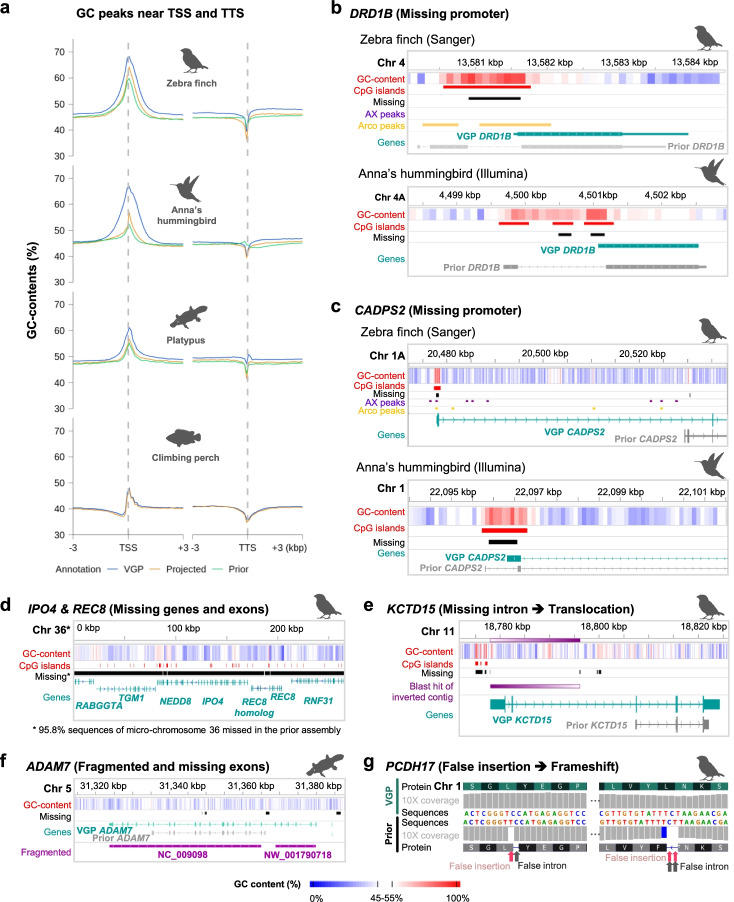
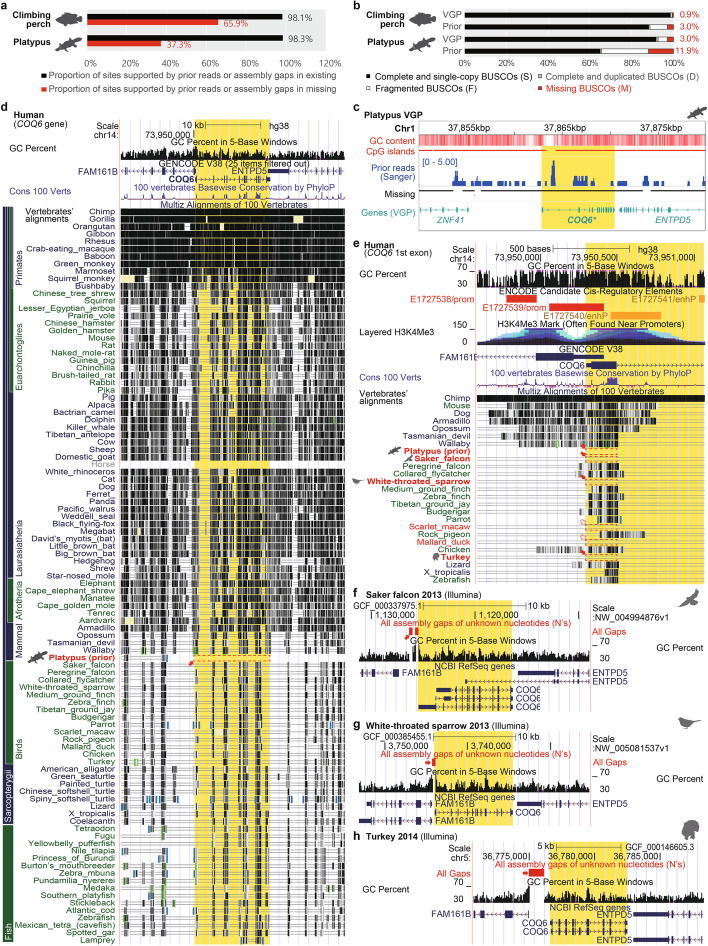
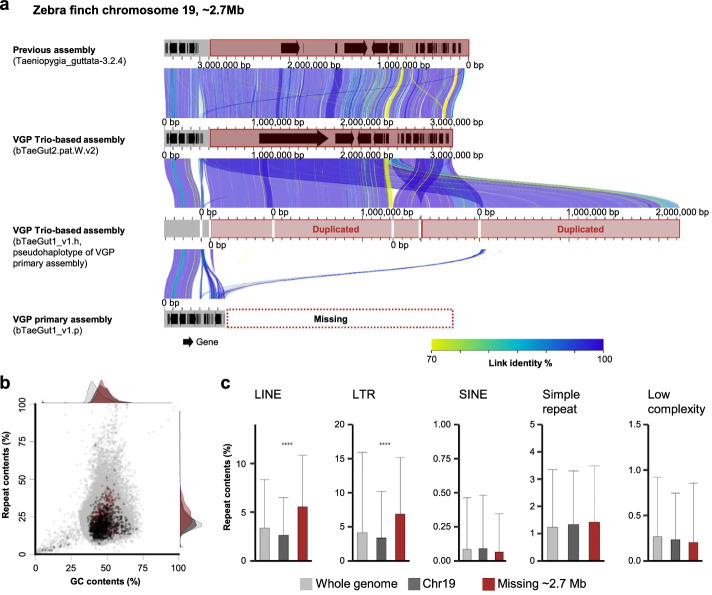
Results: Here we evaluate new vertebrate genome references relative to the previous assemblies for the same species and, in two cases, the same individuals, including a mammal (platypus), two birds (zebra finch, Anna's hummingbird), and a fish (climbing perch). We find that up to 11% of genomic sequence is entirely missing in the previous assemblies. In the Vertebrate Genomes Project zebra finch assembly, we identify eight new GC- and repeat-rich micro-chromosomes with high gene density. The impact of missing sequences is biased towards GC-rich 5'-proximal promoters and 5' exon regions of protein-coding genes and long non-coding RNAs. Between 26 and 60% of genes include structural or sequence errors that could lead to misunderstanding of their function when using the previous genome assemblies.
Conclusions: Our findings reveal novel regulatory landscapes and protein coding sequences that have been greatly underestimated in previous assemblies and are now present in the Vertebrate Genomes Project reference genomes.
Keywords: Annotation; GC content; Gene structure; Genomic dark matter; Genomics.
© 2022. The Author(s).
Conflict of interest statement
SC is currently a CEO of eGnome Inc. and GZ is currently an advisor of BGI Group: Shenzhen, Guangdong, CN. All other authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
