

Metagenome analysis using the Kraken software suite
- PMID: 36171387
- PMCID: PMC9725748
- DOI: 10.1038/s41596-022-00738-y
Metagenome analysis using the Kraken software suite
Erratum in
-
Author Correction: Metagenome analysis using the Kraken software suite.Nat Protoc. 2024 Aug 29. doi: 10.1038/s41596-024-01064-1. Online ahead of print. Nat Protoc. 2024. PMID: 39210095 No abstract available.
Abstract
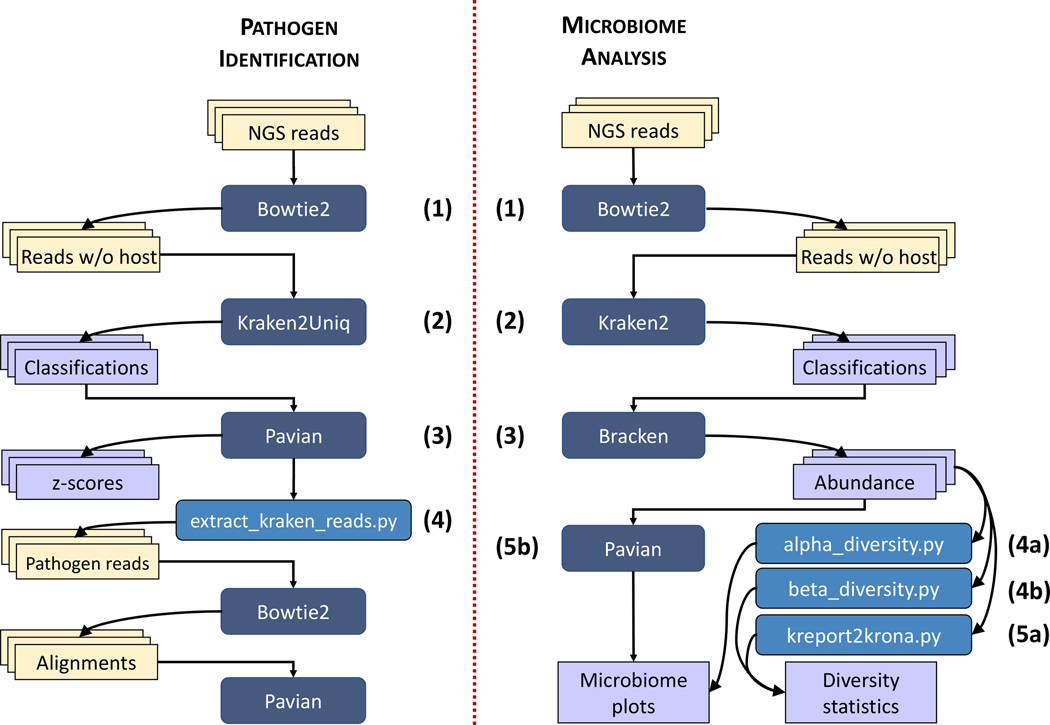
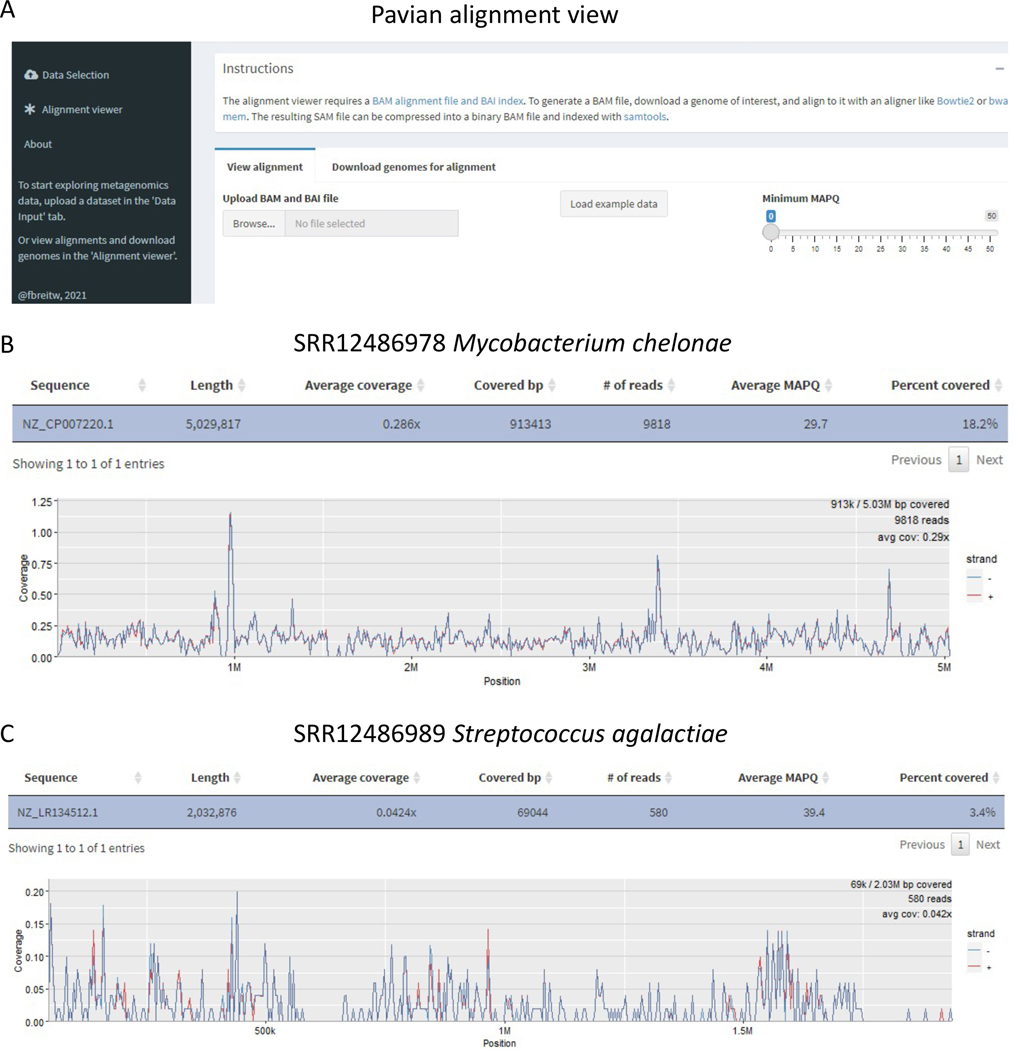
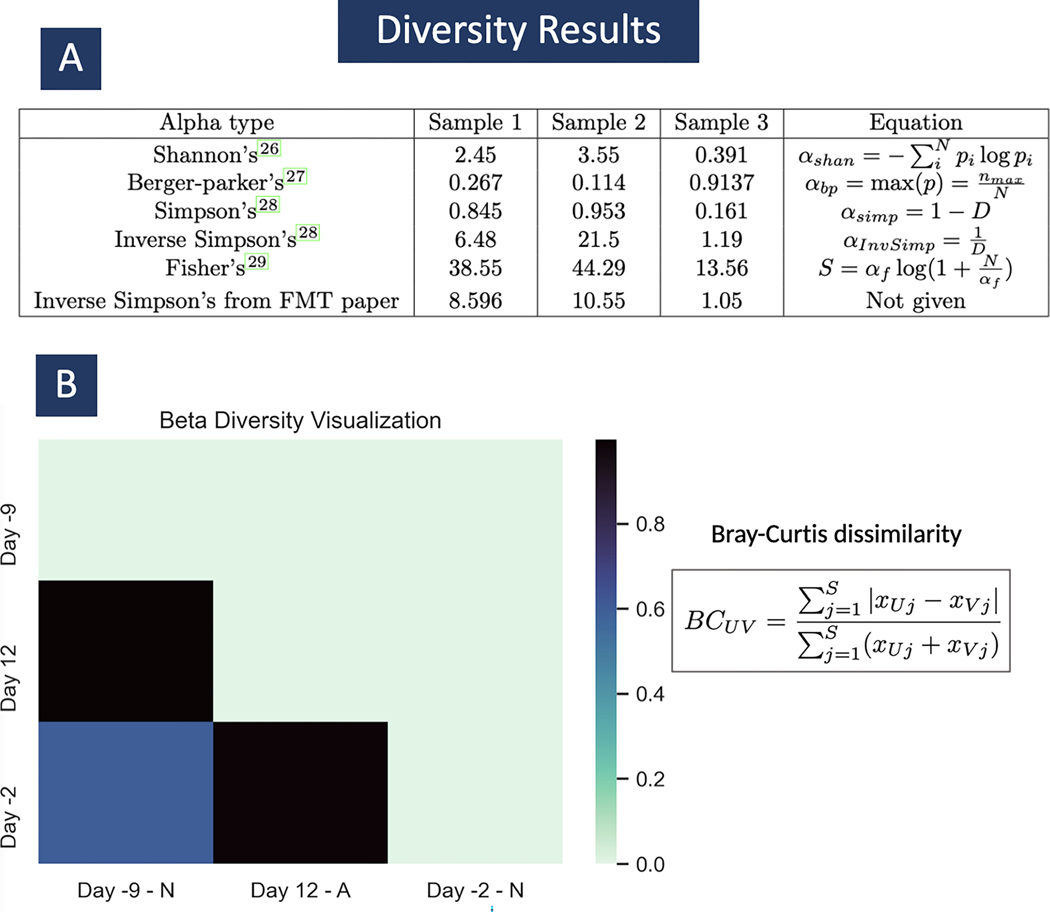
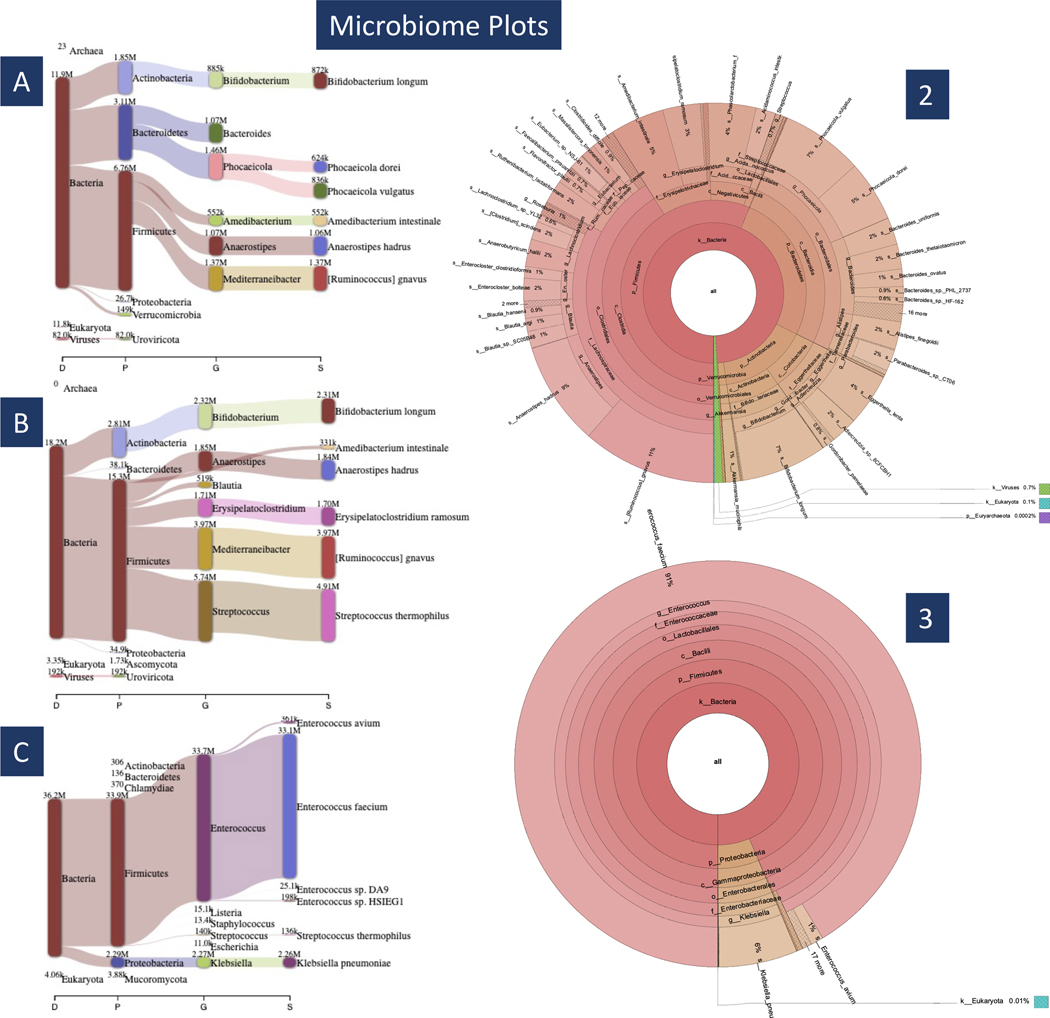
Metagenomic experiments expose the wide range of microscopic organisms in any microbial environment through high-throughput DNA sequencing. The computational analysis of the sequencing data is critical for the accurate and complete characterization of the microbial community. To facilitate efficient and reproducible metagenomic analysis, we introduce a step-by-step protocol for the Kraken suite, an end-to-end pipeline for the classification, quantification and visualization of metagenomic datasets. Our protocol describes the execution of the Kraken programs, via a sequence of easy-to-use scripts, in two scenarios: (1) quantification of the species in a given metagenomics sample; and (2) detection of a pathogenic agent from a clinical sample taken from a human patient. The protocol, which is executed within 1-2 h, is targeted to biologists and clinicians working in microbiome or metagenomics analysis who are familiar with the Unix command-line environment.
© 2022. Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing financial interest
Figures


References
-
- Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003;57:369–394. - PubMed
-
- Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci. 2017. Jan;3:e104.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
