

The three-dimensional landscape of cortical chromatin accessibility in Alzheimer's disease
- PMID: 36171428
- PMCID: PMC9581463
- DOI: 10.1038/s41593-022-01166-7
The three-dimensional landscape of cortical chromatin accessibility in Alzheimer's disease
Abstract
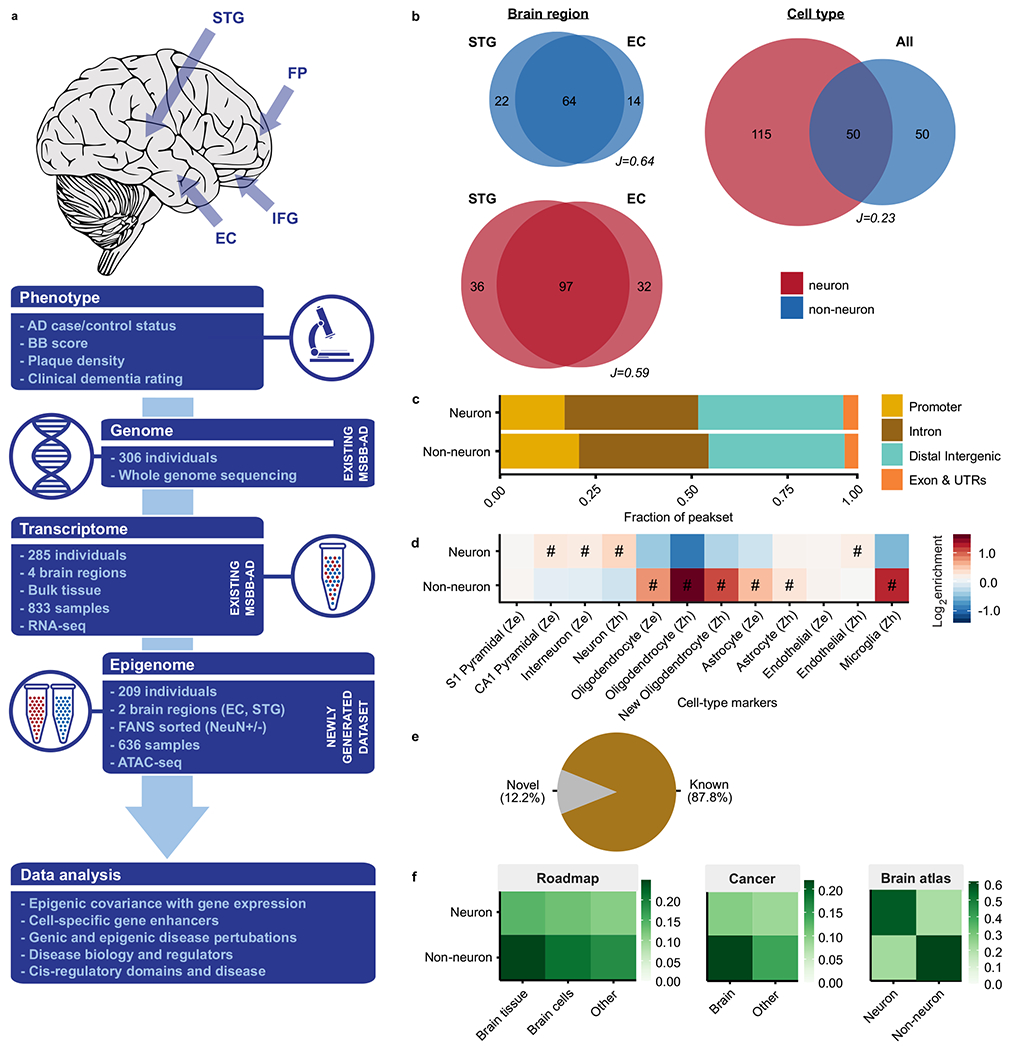
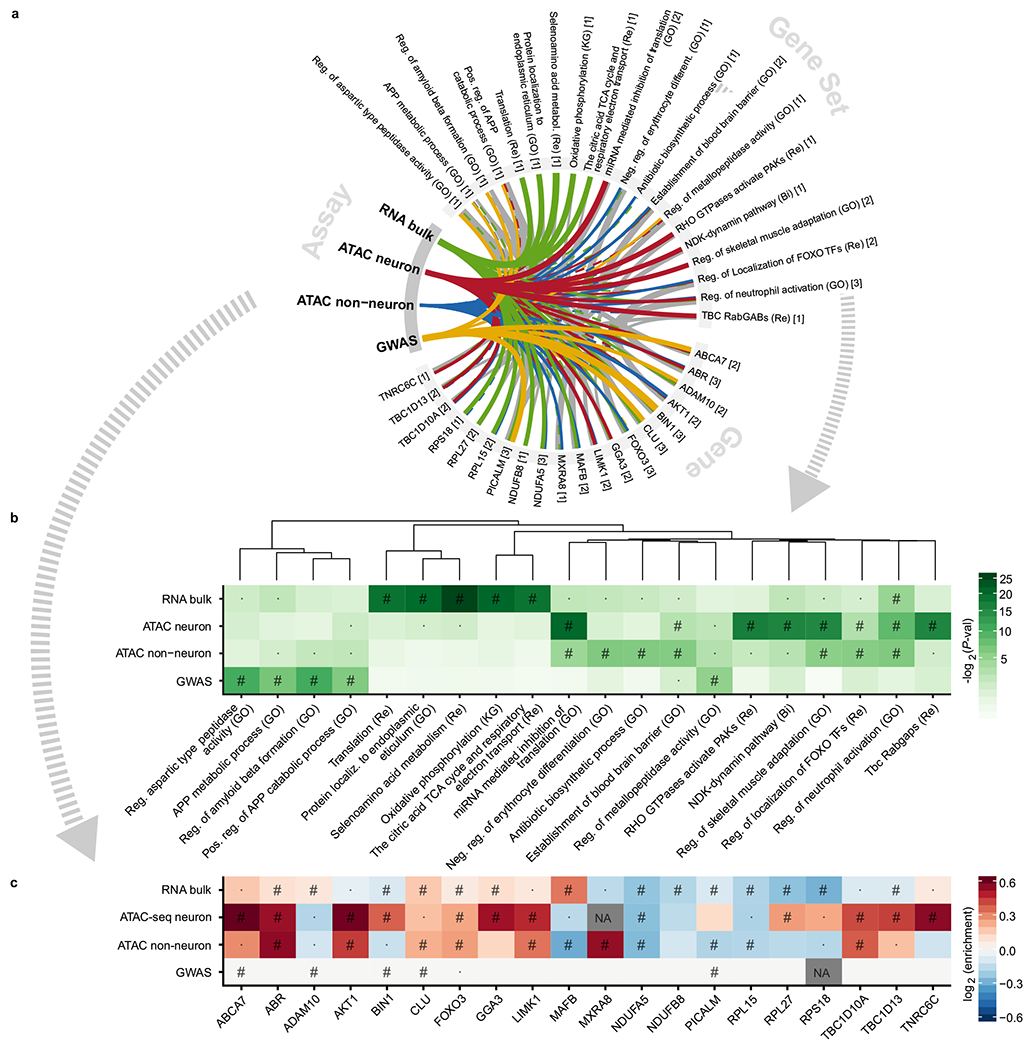
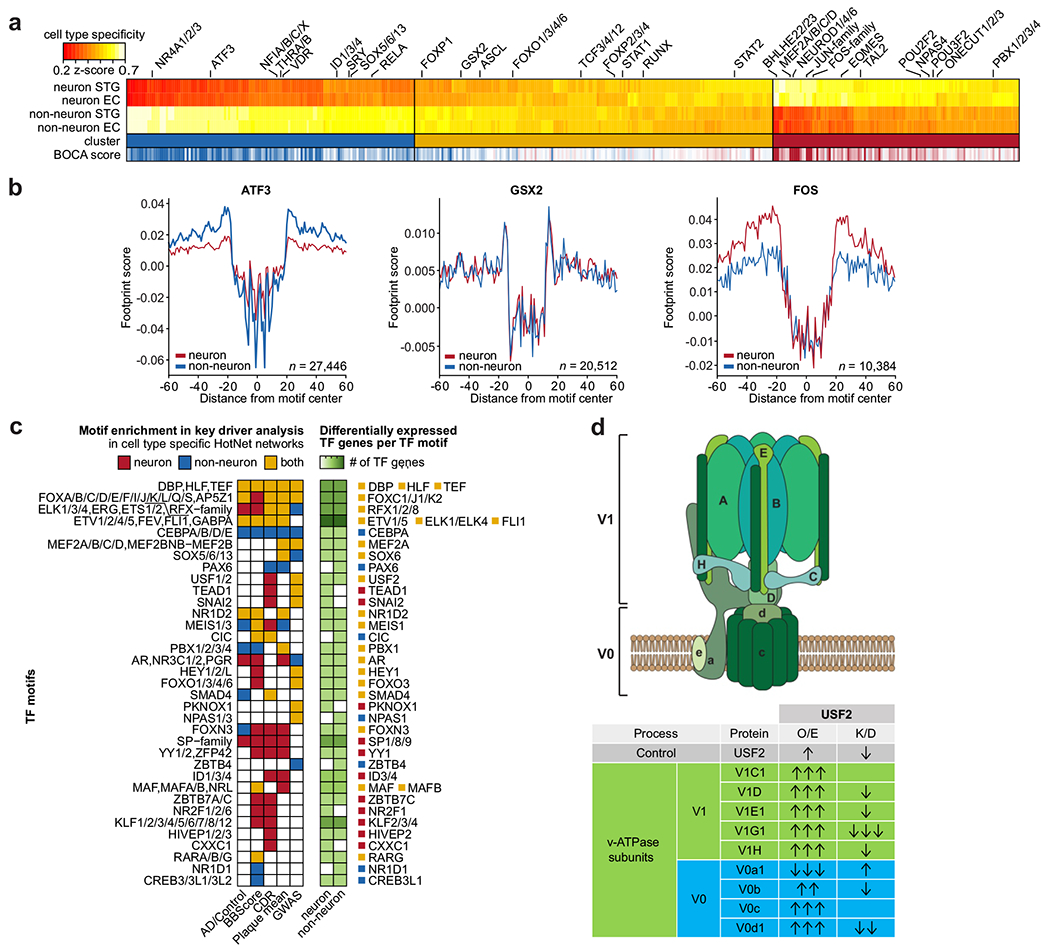
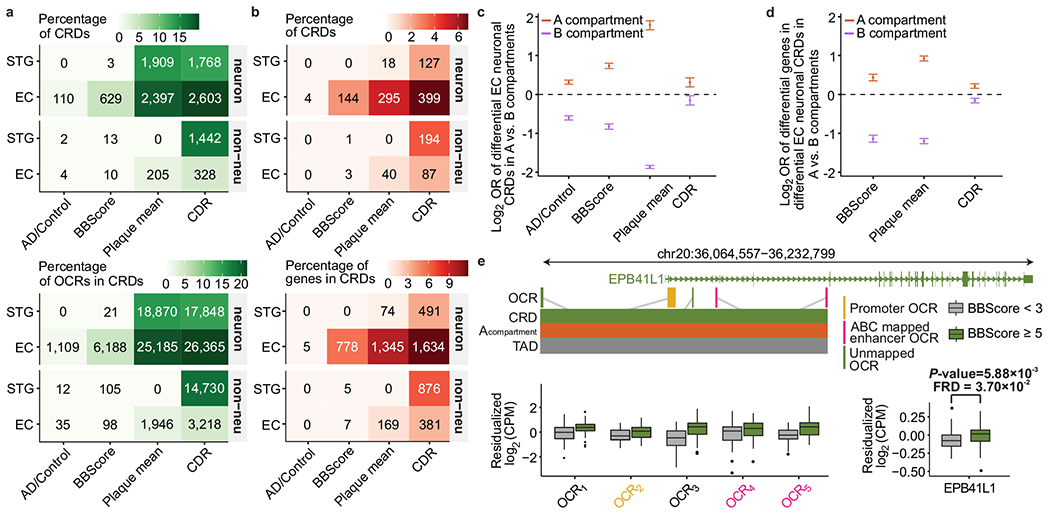
To characterize the dysregulation of chromatin accessibility in Alzheimer's disease (AD), we generated 636 ATAC-seq libraries from neuronal and nonneuronal nuclei isolated from the superior temporal gyrus and entorhinal cortex of 153 AD cases and 56 controls. By analyzing a total of ~20 billion read pairs, we expanded the repertoire of known open chromatin regions (OCRs) in the human brain and identified cell-type-specific enhancer-promoter interactions. We show that interindividual variability in OCRs can be leveraged to identify cis-regulatory domains (CRDs) that capture the three-dimensional structure of the genome (3D genome). We identified AD-associated effects on chromatin accessibility, the 3D genome and transcription factor (TF) regulatory networks. For one of the most AD-perturbed TFs, USF2, we validated its regulatory effect on lysosomal genes. Overall, we applied a systematic approach to understanding the role of the 3D genome in AD. We provide all data as an online resource for widespread community-based analysis.
© 2022. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Figures




References
-
- Marzi SJ et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci 21, 1618–1627 (2018). - PubMed
Methods-only references
-
- Morris JC et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 39, 1159–1165 (1989). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
