

Singular value decomposition of protein sequences as a method to visualize sequence and residue space
- PMID: 36173173
- PMCID: PMC9514065
- DOI: 10.1002/pro.4422
Singular value decomposition of protein sequences as a method to visualize sequence and residue space
Abstract
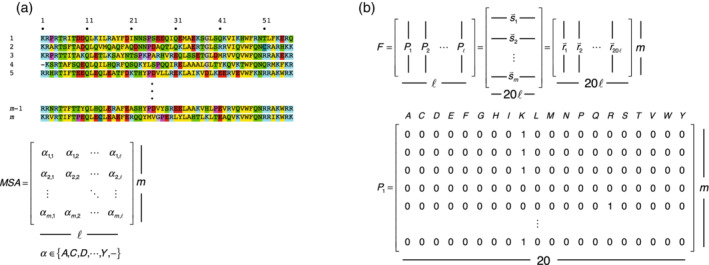
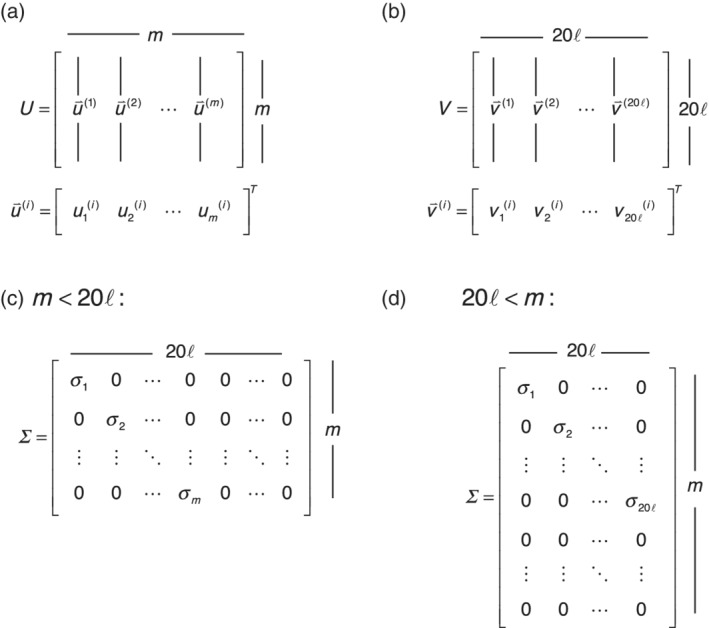
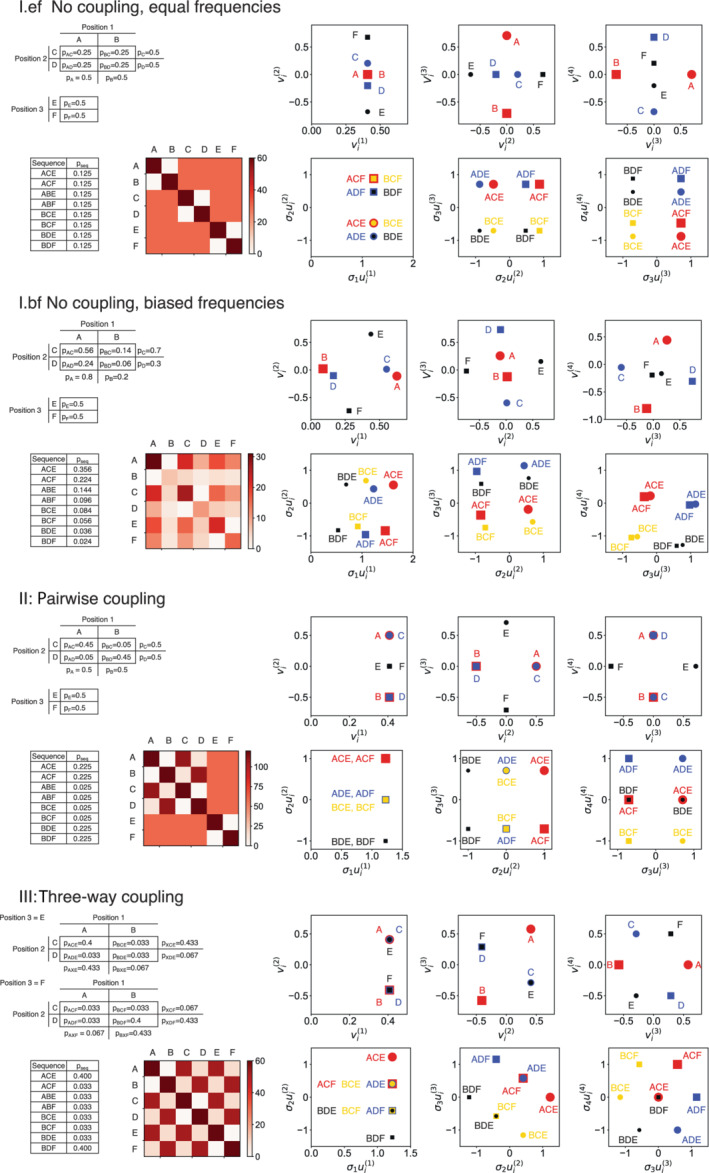
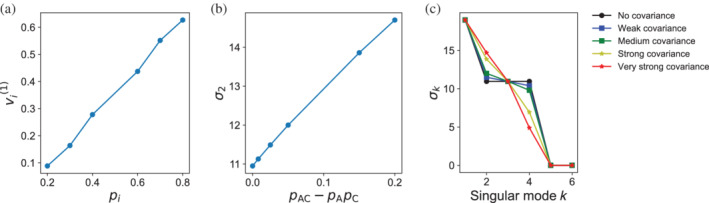
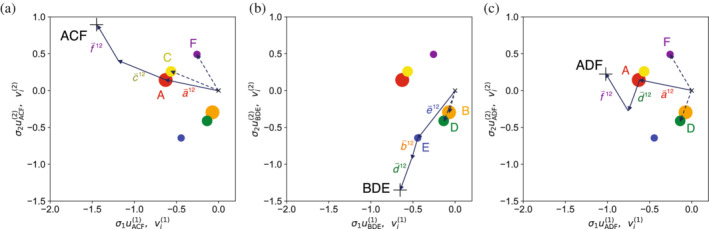
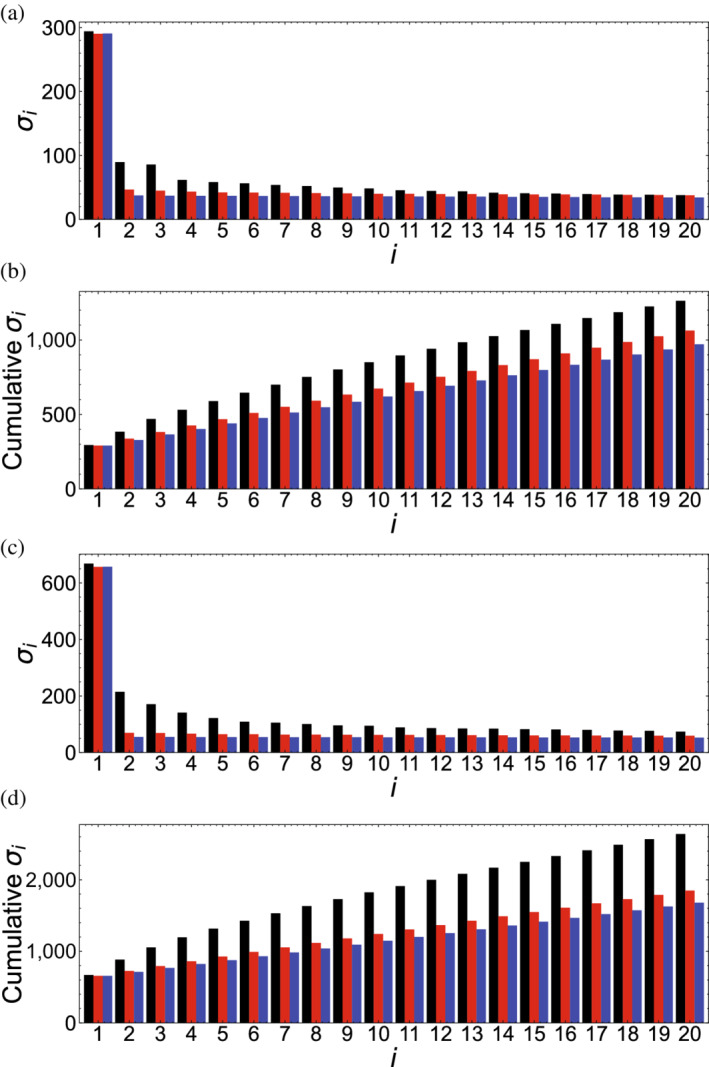
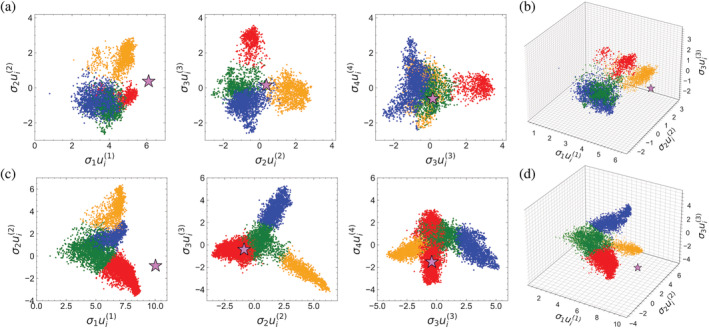
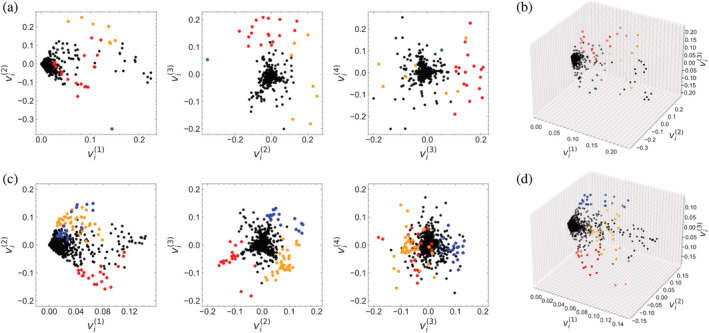
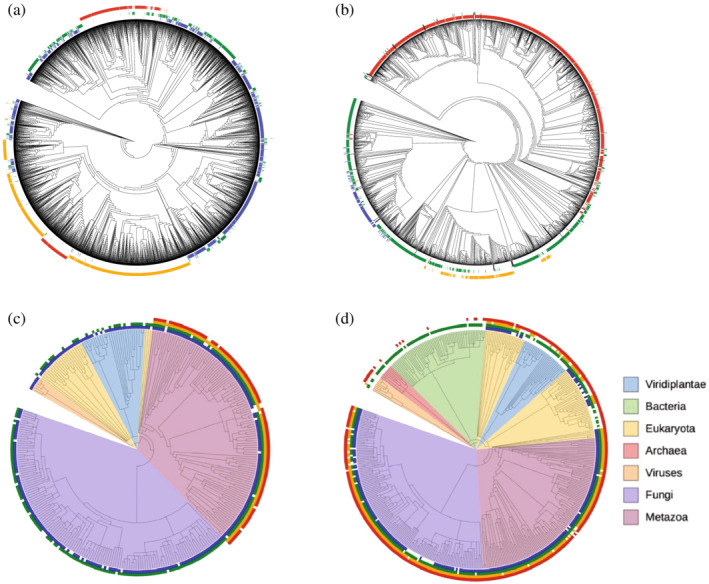
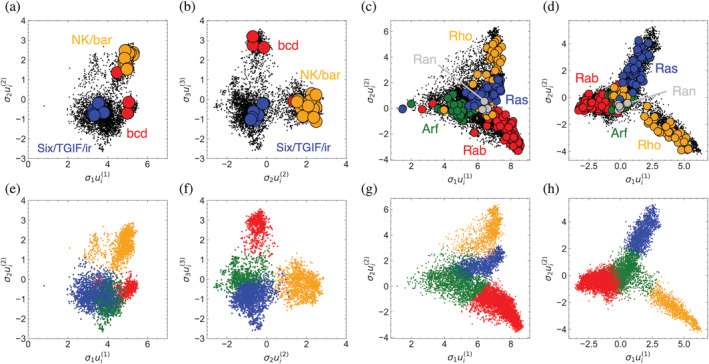
Singular value decomposition (SVD) of multiple sequence alignments (MSAs) is an important and rigorous method to identify subgroups of sequences within the MSA, and to extract consensus and covariance sequence features that define the alignment and distinguish the subgroups. This information can be correlated to structure, function, stability, and taxonomy. However, the mathematics of SVD is unfamiliar to many in the field of protein science. Here, we attempt to present an intuitive yet comprehensive description of SVD analysis of MSAs. We begin by describing the underlying mathematics of SVD in a way that is both rigorous and accessible. Next, we use SVD to analyze sequences generated with a simplified model in which the extent of sequence conservation and covariance between different positions is controlled, to show how conservation and covariance produce features in the decomposed coordinate system. We then use SVD to analyze alignments of two protein families, the homeodomain and the Ras superfamilies. Both families show clear evidence of sequence clustering when projected into singular value space. We use k-means clustering to group MSA sequences into specific clusters, show how the residues that distinguish these clusters can be identified, and show how these clusters can be related to taxonomy and function. We end by providing a description a set of Python scripts that can be used for SVD analysis of MSAs, displaying results, and identifying and analyzing sequence clusters. These scripts are freely available on GitHub.
Keywords: bioinformatics; protein design; singular value decomposition; taxonomy.
© 2022 The Protein Society.
Conflict of interest statement
The authors state no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
