

SCAFE: a software suite for analysis of transcribed cis-regulatory elements in single cells
- PMID: 36173306
- PMCID: PMC9665856
- DOI: 10.1093/bioinformatics/btac644
SCAFE: a software suite for analysis of transcribed cis-regulatory elements in single cells
Abstract
Motivation: Cell type-specific activities of cis-regulatory elements (CRE) are central to understanding gene regulation and disease predisposition. Single-cell RNA 5'end sequencing (sc-end5-seq) captures the transcription start sites (TSS) which can be used as a proxy to measure the activity of transcribed CREs (tCREs). However, a substantial fraction of TSS identified from sc-end5-seq data may not be genuine due to various artifacts, hindering the use of sc-end5-seq for de novo discovery of tCREs.
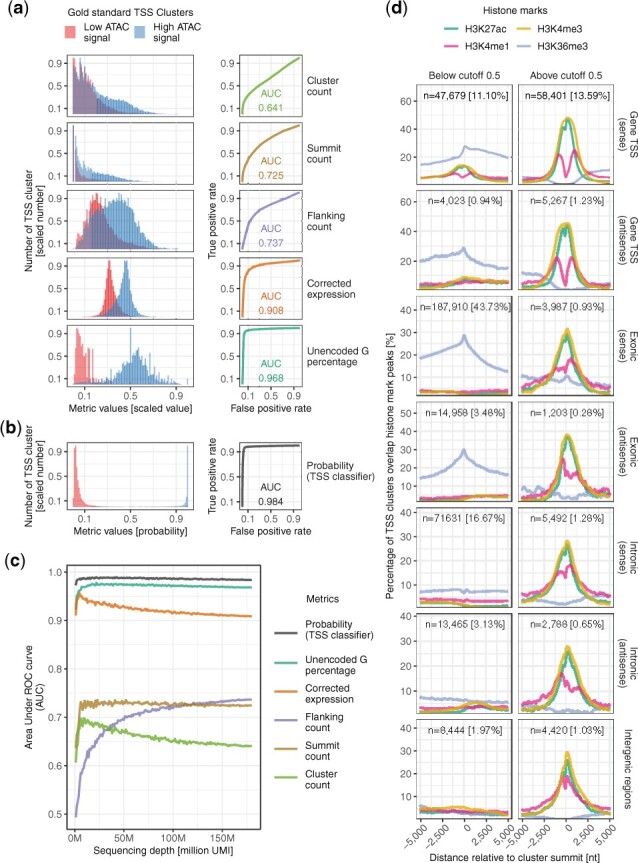
Results: We developed SCAFE-Single-Cell Analysis of Five-prime Ends-a software suite that processes sc-end5-seq data to de novo identify TSS clusters based on multiple logistic regression. It annotates tCREs based on the identified TSS clusters and generates a tCRE-by-cell count matrix for downstream analyses. The software suite consists of a set of flexible tools that could either be run independently or as pre-configured workflows.
Availability and implementation: SCAFE is implemented in Perl and R. The source code and documentation are freely available for download under the MIT License from https://github.com/chung-lab/SCAFE. Docker images are available from https://hub.docker.com/r/cchon/scafe. The submitted software version and test data are archived at https://doi.org/10.5281/zenodo.7023163 and https://doi.org/10.5281/zenodo.7024060, respectively.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press.
Figures
Similar articles
-
scGate: marker-based purification of cell types from heterogeneous single-cell RNA-seq datasets.Bioinformatics. 2022 Apr 28;38(9):2642-2644. doi: 10.1093/bioinformatics/btac141. Bioinformatics. 2022. PMID: 35258562 Free PMC article.
-
Gene count estimation with pytximport enables reproducible analysis of bulk RNA sequencing data in Python.Bioinformatics. 2024 Nov 28;40(12):btae700. doi: 10.1093/bioinformatics/btae700. Bioinformatics. 2024. PMID: 39565903 Free PMC article.
-
CAFE: a software suite for analysis of paired-sample transposon insertion sequencing data.Bioinformatics. 2021 Apr 9;37(1):121-122. doi: 10.1093/bioinformatics/btaa1086. Bioinformatics. 2021. PMID: 33393985 Free PMC article.
-
ProkSeq for complete analysis of RNA-Seq data from prokaryotes.Bioinformatics. 2021 Apr 9;37(1):126-128. doi: 10.1093/bioinformatics/btaa1063. Bioinformatics. 2021. PMID: 33367516 Free PMC article.
-
The National Rehabilitation Information Center (NARIC).J Med Libr Assoc. 2022 Jul 1;110(3):388-391. doi: 10.5195/jmla.2022.1515. J Med Libr Assoc. 2022. PMID: 36589293 Free PMC article. Review.
Cited by
-
Systematic evaluation of single-cell multimodal data integration for comprehensive human reference atlas.bioRxiv [Preprint]. 2025 Mar 6:2025.03.06.637075. doi: 10.1101/2025.03.06.637075. bioRxiv. 2025. PMID: 40093094 Free PMC article. Preprint.
-
Predicting and comparing transcription start sites in single cell populations.PLoS Comput Biol. 2025 Apr 3;21(4):e1012878. doi: 10.1371/journal.pcbi.1012878. eCollection 2025. PLoS Comput Biol. 2025. PMID: 40179341 Free PMC article.
-
CamoTSS: analysis of alternative transcription start sites for cellular phenotypes and regulatory patterns from 5' scRNA-seq data.Nat Commun. 2023 Nov 9;14(1):7240. doi: 10.1038/s41467-023-42636-1. Nat Commun. 2023. PMID: 37945584 Free PMC article.
-
Cell type-dependent directional transcription at enhancers.NAR Genom Bioinform. 2025 Mar 7;7(1):lqaf007. doi: 10.1093/nargab/lqaf007. eCollection 2025 Mar. NAR Genom Bioinform. 2025. PMID: 40060372 Free PMC article.
-
Defining the Cis-Regulatory Elements of SCN1A in GABAergic Interneurons.Mol Neurobiol. 2025 Jul 23. doi: 10.1007/s12035-025-05209-5. Online ahead of print. Mol Neurobiol. 2025. PMID: 40702288
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
