

Chronic Obstructive Pulmonary Disease and Cigarette Smoke Lead to Dysregulated Mucosal-associated Invariant T-Cell Activation
- PMID: 36174211
- PMCID: PMC9817907
- DOI: 10.1165/rcmb.2022-0131OC
Chronic Obstructive Pulmonary Disease and Cigarette Smoke Lead to Dysregulated Mucosal-associated Invariant T-Cell Activation
Abstract
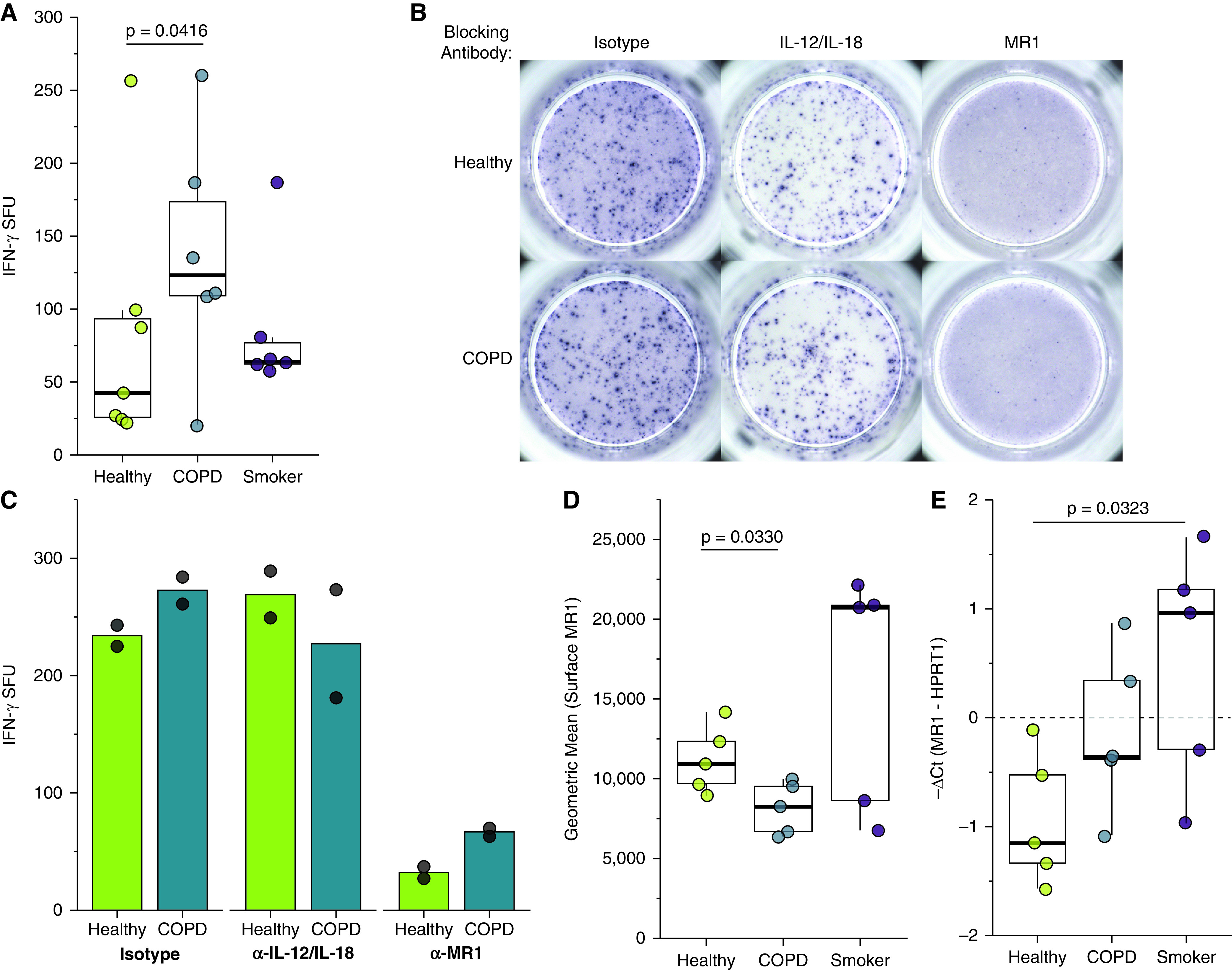
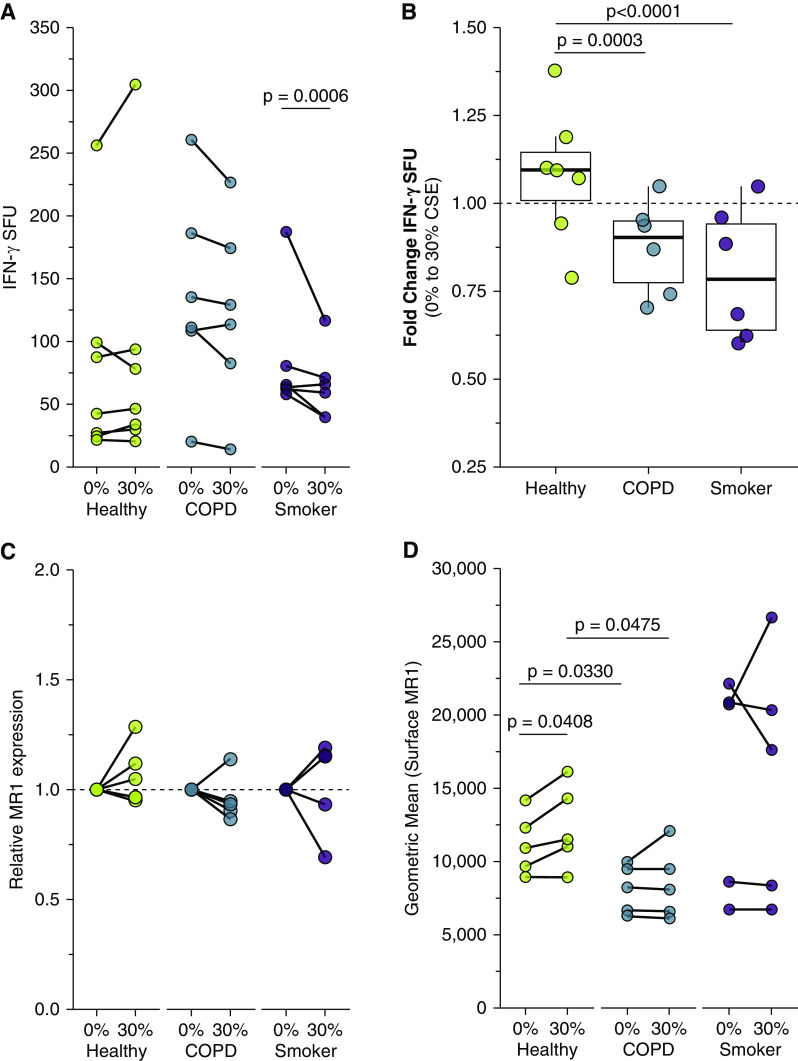
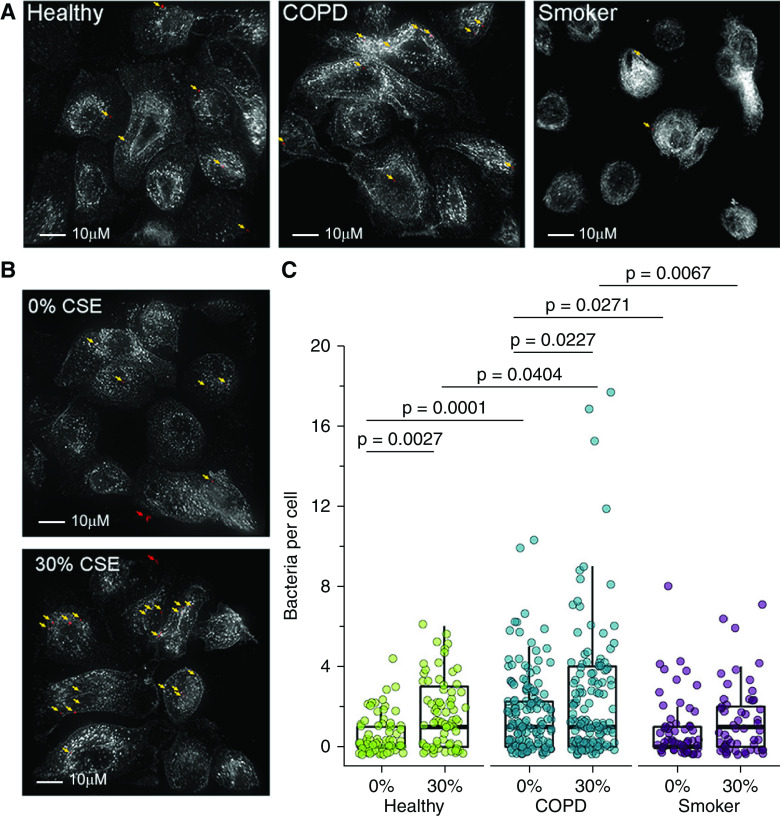
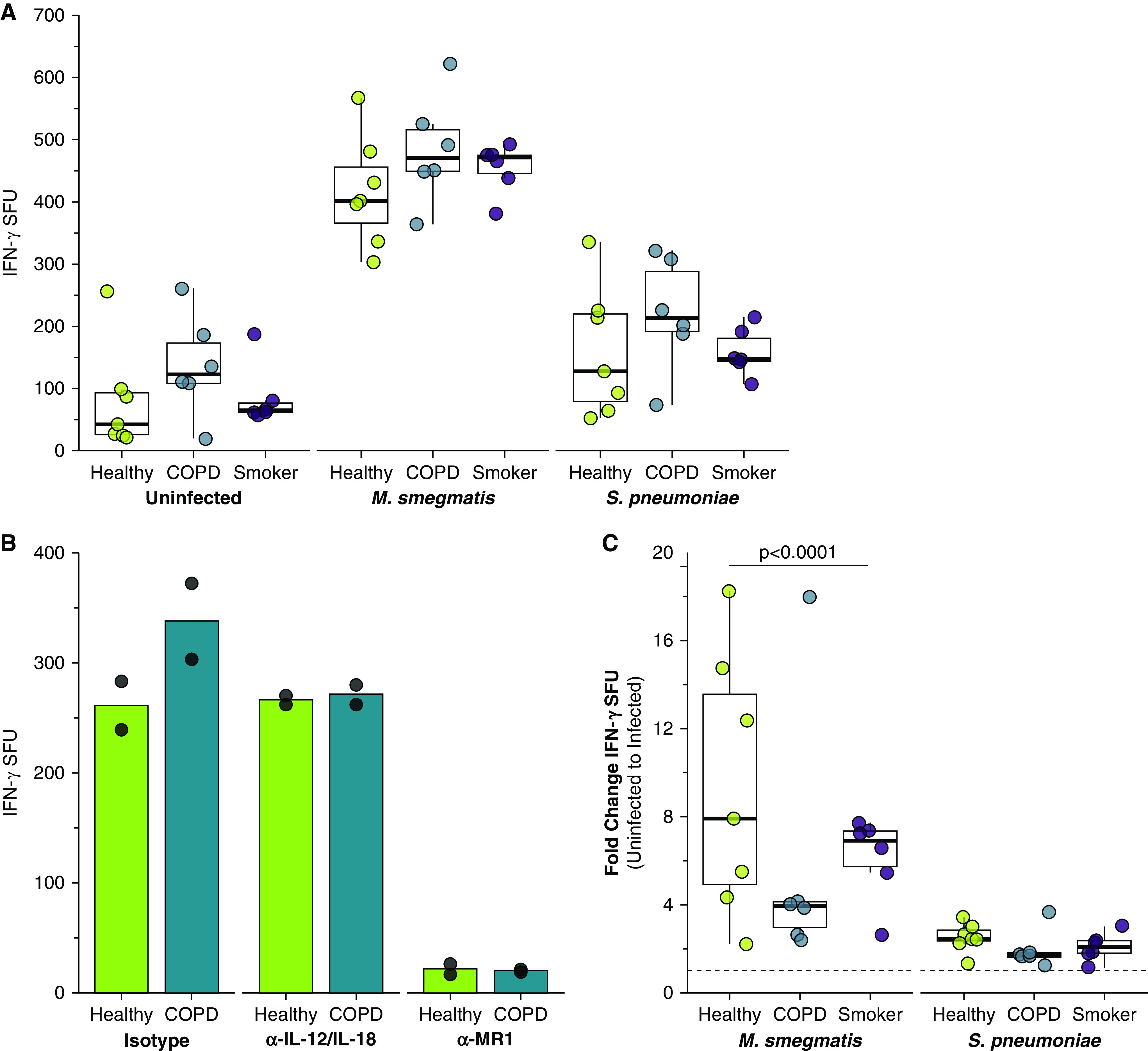
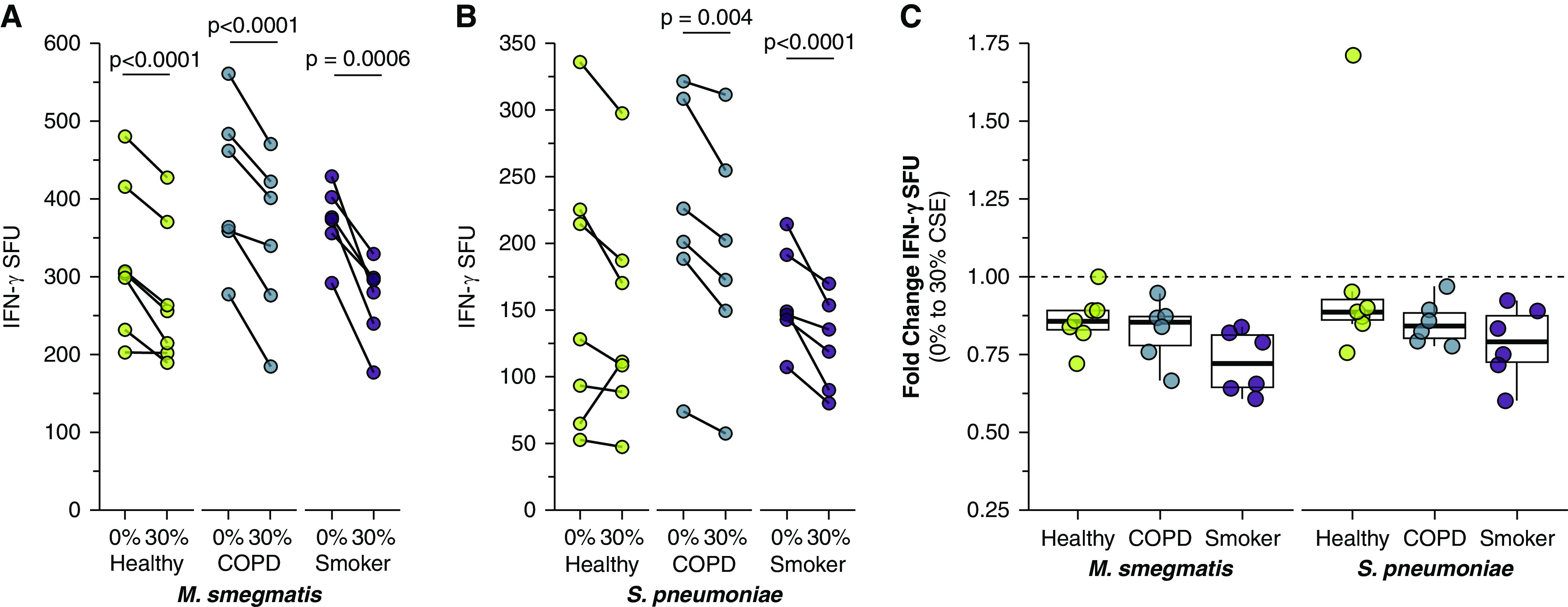
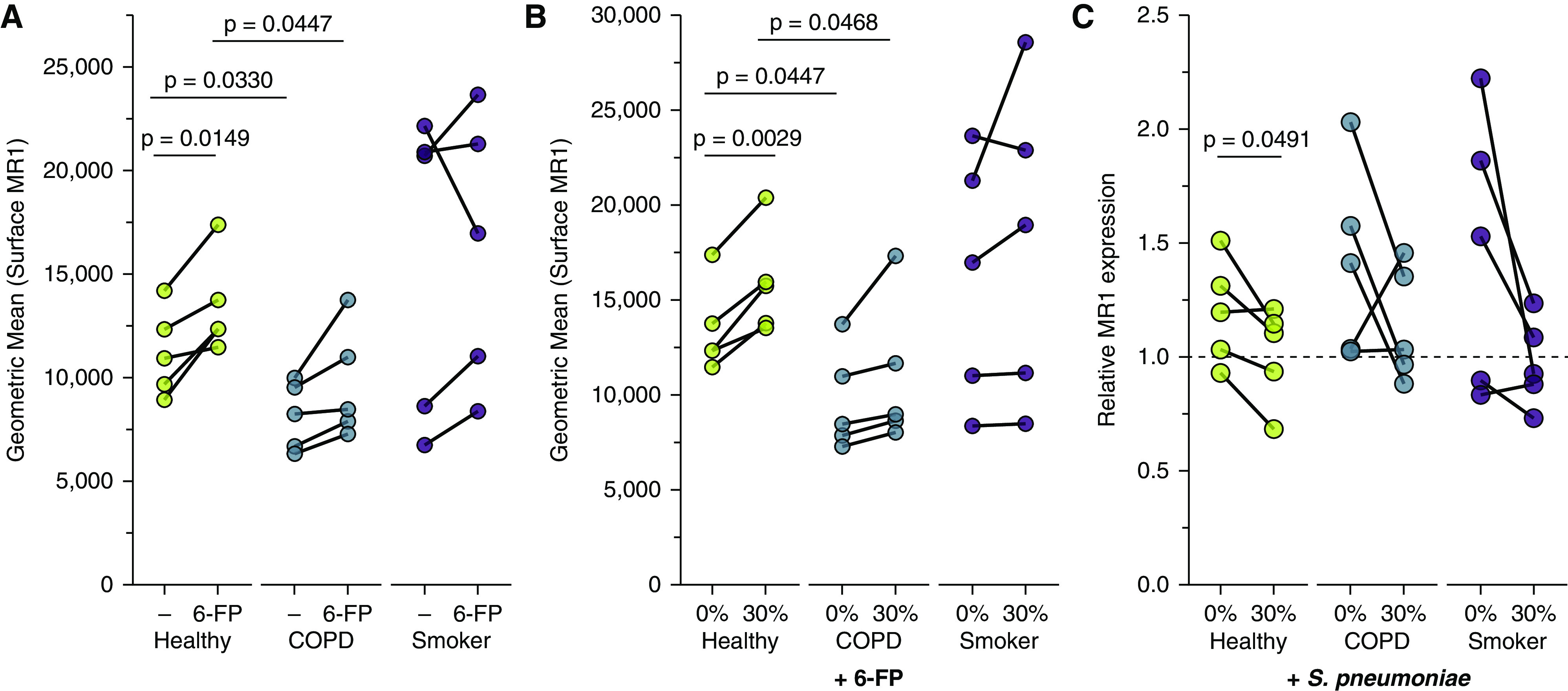
Chronic obstructive pulmonary disease (COPD) is associated with airway inflammation, increased infiltration by CD8+ T lymphocytes, and infection-driven exacerbations. Although cigarette smoke is the leading risk factor for COPD, the mechanisms driving the development of COPD in only a subset of smokers are incompletely understood. Lung-resident mucosal-associated invariant T (MAIT) cells play a role in microbial infections and inflammatory diseases. The role of MAIT cells in COPD pathology is unknown. Here, we examined MAIT cell activation in response to cigarette smoke-exposed primary human bronchial epithelial cells (BECs) from healthy, COPD, or smoker donors. We observed significantly higher baseline MAIT cell responses to COPD BECs than healthy BECs. However, infected COPD BECs stimulated a smaller fold increase in MAIT cell response despite increased microbial infection. For all donor groups, cigarette smoke-exposed BECs elicited reduced MAIT cell responses; conversely, cigarette smoke exposure increased ligand-mediated MR1 surface translocation in healthy and COPD BECs. Our data demonstrate that MAIT cell activation is dysregulated in the context of cigarette smoke and COPD. MAIT cells could contribute to cigarette smoke- and COPD-associated inflammation through inappropriate activation and reduced early recognition of bacterial infection, contributing to microbial persistence and COPD exacerbations.
Keywords: MAIT cells; MR1; Streptococcus pneumoniae; chronic obstructive pulmonary disease; cigarette smoke.
Figures
Comment in
-
Breaching the Defenses? Mucosal-associated Invariant T Cells, Smoking, and Chronic Obstructive Pulmonary Disease.Am J Respir Cell Mol Biol. 2023 Jan;68(1):9-10. doi: 10.1165/rcmb.2022-0393ED. Am J Respir Cell Mol Biol. 2023. PMID: 36256956 Free PMC article. No abstract available.
References
-
- World Health Organization. 2019.
-
- Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol . 2002;2:372–377. - PubMed
-
- Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol . 2009;9:377–384. - PubMed
-
- Nuorti JP, Butler JC, Farley MM, Harrison LH, McGeer A, Kolczak MS, et al. Active Bacterial Core Surveillance Team Cigarette smoking and invasive pneumococcal disease. N Engl J Med . 2000;342:681–689. - PubMed
-
- Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol . 2018;58:157–169. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
