

Deciphering microbial gene function using natural language processing
- PMID: 36175448
- PMCID: PMC9523054
- DOI: 10.1038/s41467-022-33397-4
Deciphering microbial gene function using natural language processing
Abstract
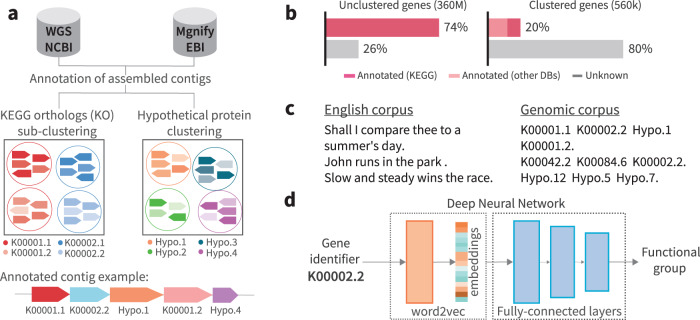
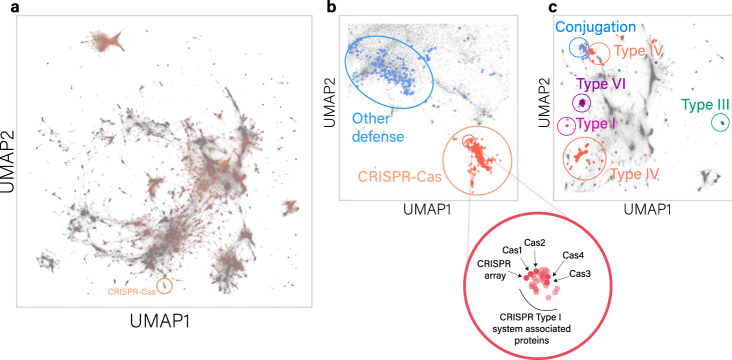
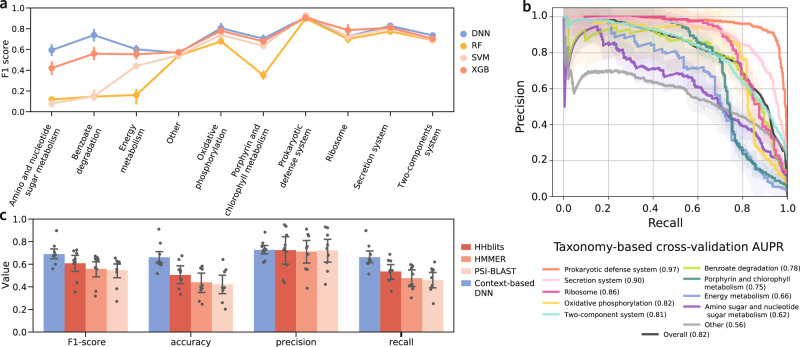
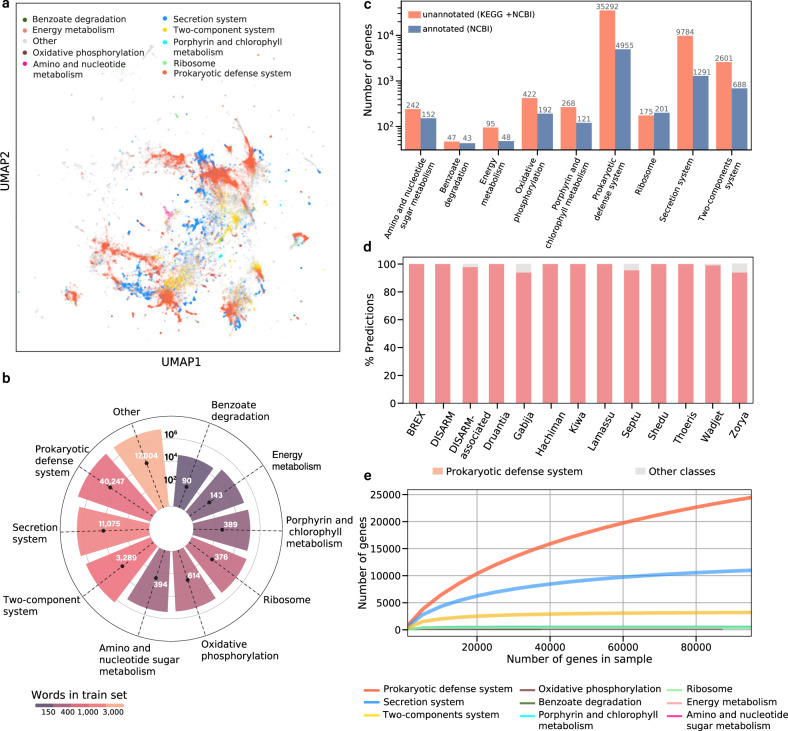
Revealing the function of uncharacterized genes is a fundamental challenge in an era of ever-increasing volumes of sequencing data. Here, we present a concept for tackling this challenge using deep learning methodologies adopted from natural language processing (NLP). We repurpose NLP algorithms to model "gene semantics" based on a biological corpus of more than 360 million microbial genes within their genomic context. We use the language models to predict functional categories for 56,617 genes and find that out of 1369 genes associated with recently discovered defense systems, 98% are inferred correctly. We then systematically evaluate the "discovery potential" of different functional categories, pinpointing those with the most genes yet to be characterized. Finally, we demonstrate our method's ability to discover systems associated with microbial interaction and defense. Our results highlight that combining microbial genomics and language models is a promising avenue for revealing gene functions in microbes.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
