

Concurrent inhibition of CDK2 adds to the anti-tumour activity of CDK4/6 inhibition in GIST
- PMID: 36175617
- PMCID: PMC9681737
- DOI: 10.1038/s41416-022-01990-5
Concurrent inhibition of CDK2 adds to the anti-tumour activity of CDK4/6 inhibition in GIST
Abstract
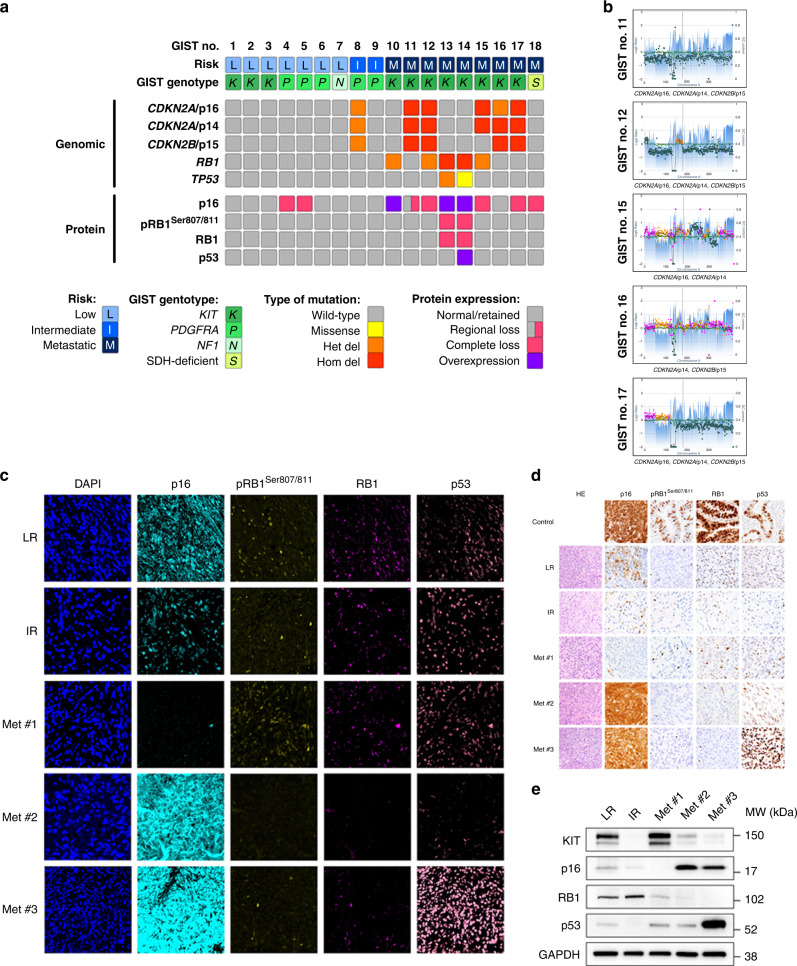
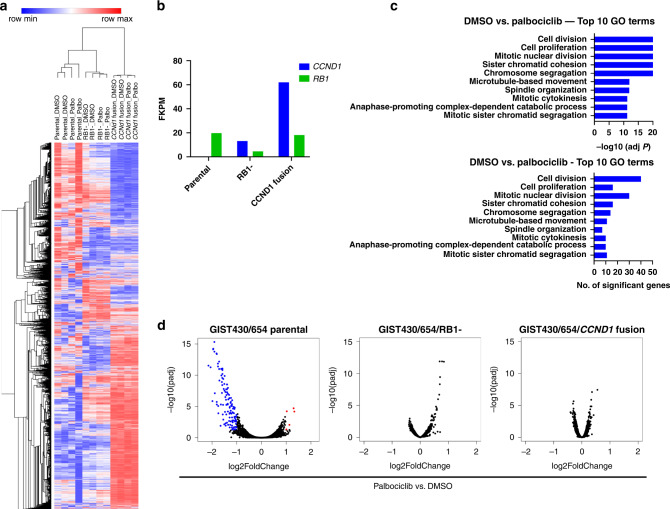
Background: Advanced gastrointestinal stromal tumour (GIST) is characterised by genomic perturbations of key cell cycle regulators. Oncogenic activation of CDK4/6 results in RB1 inactivation and cell cycle progression. Given that single-agent CDK4/6 inhibitor therapy failed to show clinical activity in advanced GIST, we evaluated strategies for maximising response to therapeutic CDK4/6 inhibition.
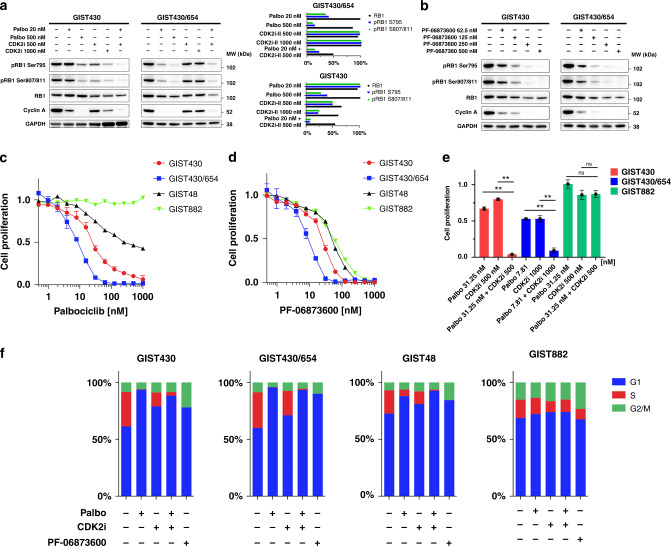
Methods: Targeted next-generation sequencing and multiplexed protein imaging were used to detect cell cycle regulator aberrations in GIST clinical samples. The impact of inhibitors of CDK2, CDK4 and CDK2/4/6 was determined through cell proliferation and protein detection assays. CDK-inhibitor resistance mechanisms were characterised in GIST cell lines after long-term exposure.
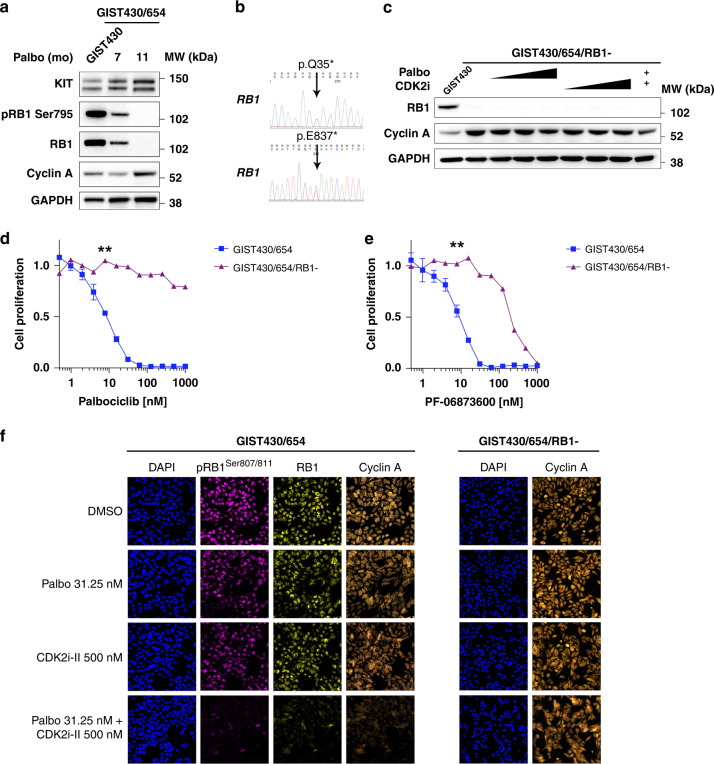
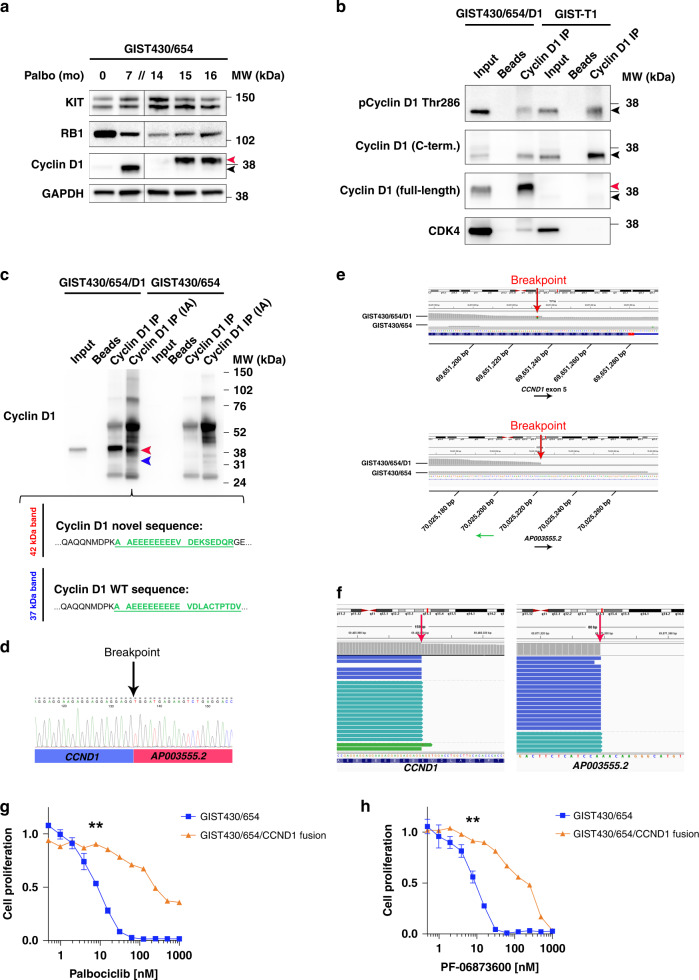
Results: We identify recurrent genomic aberrations in cell cycle regulators causing co-activation of the CDK2 and CDK4/6 pathways in clinical GIST samples. Therapeutic co-targeting of CDK2 and CDK4/6 is synergistic in GIST cell lines with intact RB1, through inhibition of RB1 hyperphosphorylation and cell proliferation. Moreover, RB1 inactivation and a novel oncogenic cyclin D1 resulting from an intragenic rearrangement (CCND1::chr11.g:70025223) are mechanisms of acquired CDK-inhibitor resistance in GIST.
Conclusions: These studies establish the biological rationale for CDK2 and CDK4/6 co-inhibition as a therapeutic strategy in patients with advanced GIST, including metastatic GIST progressing on tyrosine kinase inhibitors.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
SKS and PY are inventors for a US patent application for the multiplexed imaging technology used in this work. PY is a co-founder of Ultivue, Inc. and NuProbe Global. SG serves as consultant/advisory board member to Deciphera Pharmaceuticals, Blueprint Medicines, Daiichi-Sankyo, Kayothera, Immunicum, Eli Lilly, Bayer, Ayala; reports research funding to the institution by Deciphera Pharmaceuticals, Blueprint Medicines, Daiichi-Sankyo, Theseus Pharmaceuticals, Merck, Eisai, Springworks, Pfizer and Bayer; holds equity at Abbott Laboratories; and receives royalties from Wolters Kluwer/UpToDate. MMB serves on the Board of Directors of Natera, Inc., and Leap Therapeutics. JLH serves as a consultant to Aadi Biosciences and TRACON Pharmaceuticals. GDD serves as a Board of Directors member with minor equity holding in Blueprint Medicines; serves as co-founder with minor equity holding in IDRX; serves as consultant/SAB member with minor equity holding in G1 Therapeutics, Caris Life Sciences, Erasca Pharmaceuticals, RELAY Therapeutics, Bessor Pharmaceuticals, CellCarta, IKENA Oncology, Kojin Therapeutics, Acrivon Therapeutics; serves as a scientific consultant with sponsored research to Dana-Farber to Bayer, Pfizer, Novartis, Roche/Genentech, Janssen, PharmaMar, Daiichi-Sankyo, AdaptImmune; serves as a scientific consultant to GlaxoSmithKline, EMD-Serono, MEDSCAPE, Mirati, WCG/Arsenal Capital, RAIN Therapeutics; and receives Novartis royalty to Dana-Farber for use patent of imatinib in GIST. The remaining authors declare no competing interests.
Figures

References
-
- Heinrich MC, Rankin C, Blanke CD, Demetri GD, Borden EC, Ryan CW, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of phase 3 SWOG intergroup trial S0033. JAMA Oncol. 2017;3:944–52. doi: 10.1001/jamaoncol.2016.6728. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
