

Genetic Evaluation of A Nation-Wide Dutch Pediatric DCM Cohort: The Use of Genetic Testing in Risk Stratification
- PMID: 36178741
- PMCID: PMC9622377
- DOI: 10.1161/CIRCGEN.120.002981
Genetic Evaluation of A Nation-Wide Dutch Pediatric DCM Cohort: The Use of Genetic Testing in Risk Stratification
Abstract
Background: This study aimed to describe the current practice and results of genetic evaluation in Dutch children with dilated cardiomyopathy and to evaluate genotype-phenotype correlations that may guide prognosis.
Methods: We performed a multicenter observational study in children diagnosed with dilated cardiomyopathy, from 2010 to 2017.
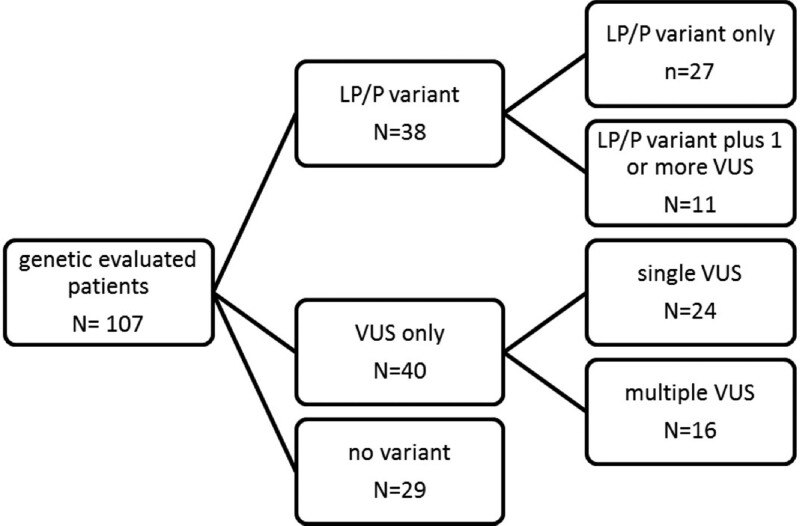
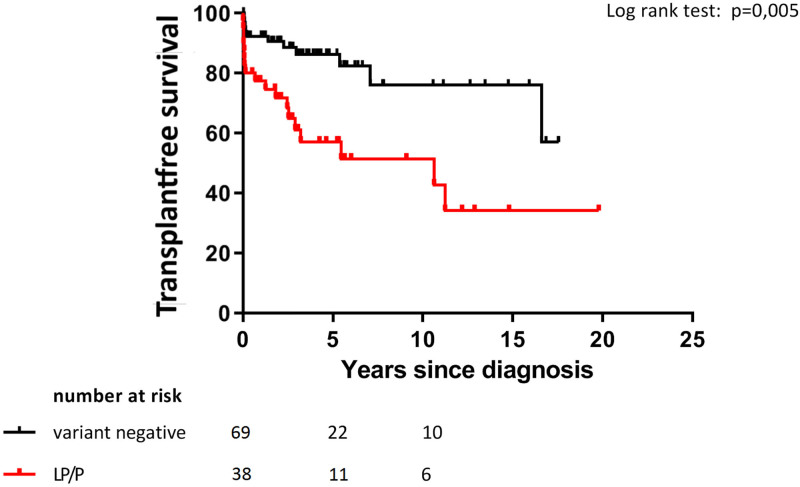
Results: One hundred forty-four children were included. Initial diagnostic categories were idiopathic dilated cardiomyopathy in 67 children (47%), myocarditis in 23 (16%), neuromuscular in 7 (5%), familial in 18 (13%), inborn error of metabolism in 4 (3%), malformation syndrome in 2 (1%), and "other" in 23 (16%). Median follow-up time was 2.1 years [IQR 1.0-4.3]. Hundred-seven patients (74%) underwent genetic testing. We found a likely pathogenic or pathogenic variant in 38 children (36%), most often in MYH7 (n = 8). In 1 patient initially diagnosed with myocarditis, a pathogenic LMNA variant was found. During the study, 39 patients (27%) reached study endpoint (SE: all-cause death or heart transplantation). Patients with a likely pathogenic or pathogenic variant were more likely to reach SE compared with those without (hazard ratio 2.8; 95% CI 1.3-5.8, P = 0.007), while transplant-free survival was significantly lower (P = 0.006). Clinical characteristics at diagnosis did not differ between the 2 groups.
Conclusions: Genetic testing is a valuable tool for predicting prognosis in children with dilated cardiomyopathy, with carriers of a likely pathogenic or pathogenic variant having a worse prognosis overall. Genetic testing should be incorporated in clinical work-up of all children with dilated cardiomyopathy regardless of presumed disease pathogenesis.
Keywords: cardiomyopathy, dilated; genetic testing; pediatric cardiology.
Figures
References
-
- Olson TM, Kishimoto NY, Whitby FG, Michels VV. Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol. 2001;33:723–732. doi: 10.1006/jmcc.2000.1339 - PubMed
-
- Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280:750–752. doi: 10.1126/science.280.5364.750 - PubMed
-
- Towbin JA, Bowles NE. Genetic abnormalities responsible for dilated cardiomyopathy. Curr Cardiol Rep. 2000;2:475–480. doi: 10.1007/s11886-000-0063-9 - PubMed
-
- Harakalova M, Kummeling G, Sammani A, Linschoten M, Baas AF, van der Smagt J, Doevendans PA, van Tintelen JP, Dooijes D, Mokry M, et al. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific. Eur J Heart Fail. 2015;17:484–493. doi: 10.1002/ejhf.255 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
