

Evolution of SARS-CoV-2 during the first year of the COVID-19 pandemic in Northwestern Argentina
- PMID: 36181975
- PMCID: PMC9599208
- DOI: 10.1016/j.virusres.2022.198936
Evolution of SARS-CoV-2 during the first year of the COVID-19 pandemic in Northwestern Argentina
Abstract
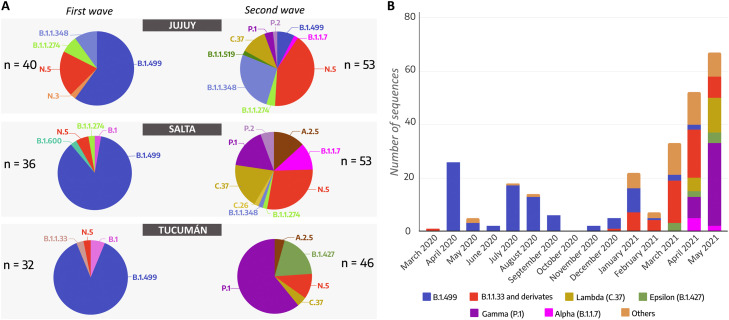
Studies about the evolution of SARS-CoV-2 lineages in different backgrounds such as naive populations are still scarce, especially from South America. This work aimed to study the introduction and diversification pattern of SARS-CoV-2 during the first year of the COVID-19 pandemic in the Northwestern Argentina (NWA) region and to analyze the evolutionary dynamics of the main lineages found. In this study, we analyzed a total of 260 SARS-CoV-2 whole-genome sequences from Argentina, belonging to the Provinces of Jujuy, Salta, and Tucumán, from March 31st, 2020, to May 22nd, 2021, which covered the full first wave and the early second wave of the COVID-19 pandemic in Argentina. In the first wave, eight lineages were identified: B.1.499 (76.9%), followed by N.5 (10.2%), B.1.1.274 (3.7%), B.1.1.348 (3.7%), B.1 (2.8%), B.1.600 (0.9%), B.1.1.33 (0.9%) and N.3 (0.9%). During the early second wave, the first-wave lineages were displaced by the introduction of variants of concern (VOC) (Alpha, Gamma), or variants of interest (VOI) (Lambda, Zeta, Epsilon) and other lineages with more limited distribution. Phylodynamic analyses of the B.1.499 and N.5, the two most prevalent lineages in the NWA, revealed that the rate of evolution of lineage N.5 (7.9 × 10-4 substitutions per site per year, s/s/y) was a ∼40% faster than that of lineage B.1.499 (5.6 × 10-4 s/s/y), although both are in the same order of magnitude than other non-VOC lineages. No mutations associated with a biological characteristic of importance were observed as signatures markers of the phylogenetic groups established in Northwestern Argentina, however, single sequences in non-VOC lineages did present mutations of biological importance or associated with VOCs as sporadic events, showing that many of these mutations could emerge from circulation in the general population. This study contributed to the knowledge about the evolution of SARS-CoV-2 in a pre-vaccination and without post-exposure immunization period.
Keywords: Argentina; COVID-19; First-wave lineages; Rate of evolution; SARS-CoV-2.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures










References
LinkOut - more resources
Full Text Sources
Miscellaneous
