

Three highly variable genome regions of the four dengue virus serotypes can accurately recapitulate the CDS phylogeny
- PMID: 36187156
- PMCID: PMC9516459
- DOI: 10.1016/j.mex.2022.101859
Three highly variable genome regions of the four dengue virus serotypes can accurately recapitulate the CDS phylogeny
Abstract
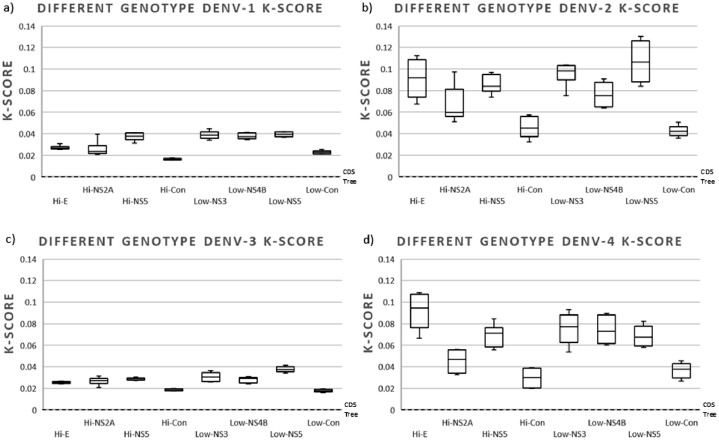
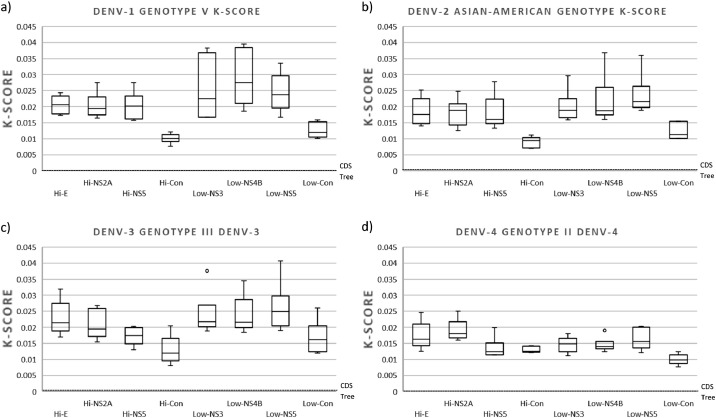
The circulation of the four-dengue virus (DENV) serotypes has significantly increased in recent years, accompanied by an increase in viral genetic diversity. In order to conduct disease surveillance and understand DENV evolution and its effects on virus transmission and disease, efficient and accurate methods for phylogenetic classification are required. Phylogenetic analysis of different viral genes sequences is the most used method, the envelope gene (E) being the most frequently selected target. We explored the genetic variability of the four DENV serotypes throughout their complete coding sequence (CDS) of sequences available in GenBank and used genomic regions of different variability rate to recapitulate the phylogeny obtained with the DENV CDS. Our results indicate that the use of high or low variable regions accurately recapitulate the phylogeny obtained with CDS of sequences from different DENV genotypes. However, when analyzing the phylogeny of a single genotype, highly variable regions performed better in recapitulating the distance branch length, topology, and support of the CDS phylogeny. The use of three concatenated highly variable regions was not statistically different in distance branch length and support to that obtained in CDS phylogeny.•This study demonstrated the ability of highly variable regions of the DENV genome to recapitulate the phylogeny obtained with the full coding sequence (CDS).•The use of genomic regions of high or low variability did not affect the performance in recapitulating the phylogeny obtained with CDS from different genotypes. However, when phylogeny was analyzed for sequences from a single genotype, highly variable regions performed better in recapitulating the distance branch length, topology, and support of the CDS phylogeny.•The use of concatenated highly variable genome regions represent a useful option for recapitulating genome-wide phylogenies in analyses of sequences belonging to the same DENV genotype.
Keywords: Bayesian inference; Dengue virus; Genotyping; Highly variable regions; Lineages; Phylogeny.
© 2022 The Author(s). Published by Elsevier B.V.
Figures








References
-
- Parameswaran P., Charlebois P., Tellez Y., Nunez A., Ryan E.M., Malboeuf C.M., Levin J.Z., Lennon N.J., Balmaseda A., Harris E., Henn M.R. Genome-wide patterns of intrahuman dengue virus diversity reveal associations with viral phylogenetic clade and interhost diversity. J Virol. 2012;86:8546–8558. doi: 10.1128/JVI.00736-12. - DOI - PMC - PubMed
-
- Holmes E.C., Twiddy S.S. The origin, emergence and evolutionary genetics of dengue virus. Infect Genet Evol. 2003;3:19–28. - PubMed
LinkOut - more resources
Full Text Sources
