

Blau syndrome NOD2 mutations result in loss of NOD2 cross-regulatory function
- PMID: 36189261
- PMCID: PMC9520668
- DOI: 10.3389/fimmu.2022.988862
Blau syndrome NOD2 mutations result in loss of NOD2 cross-regulatory function
Abstract
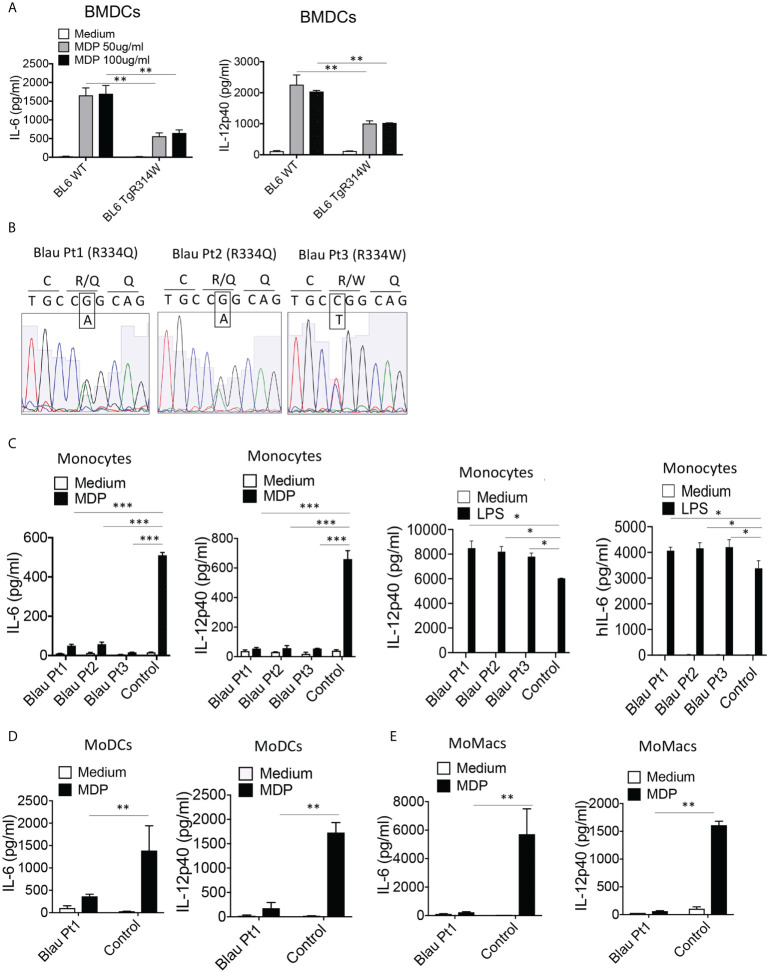
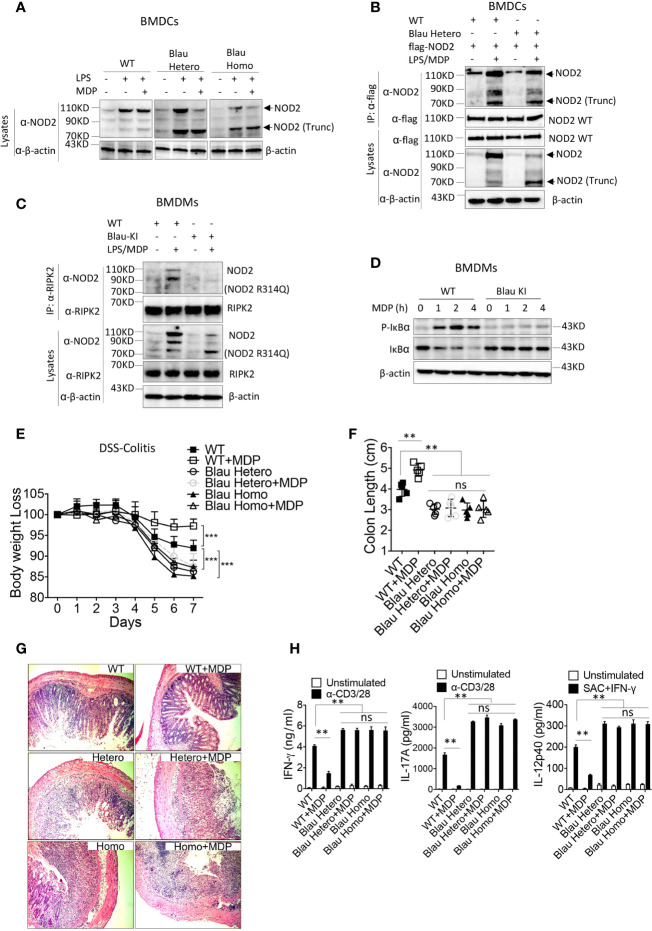
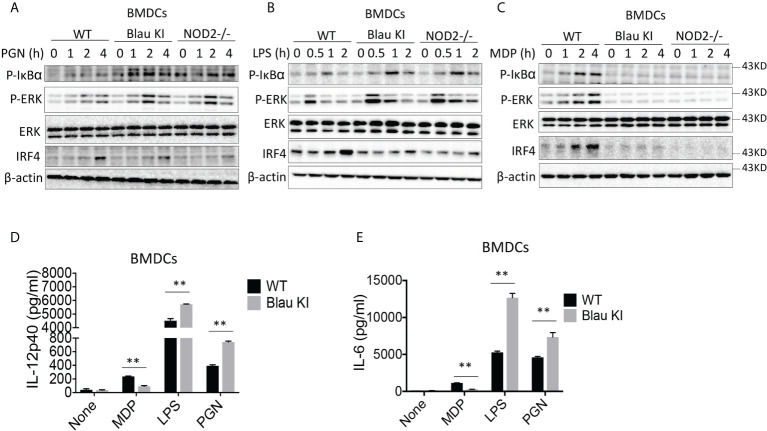
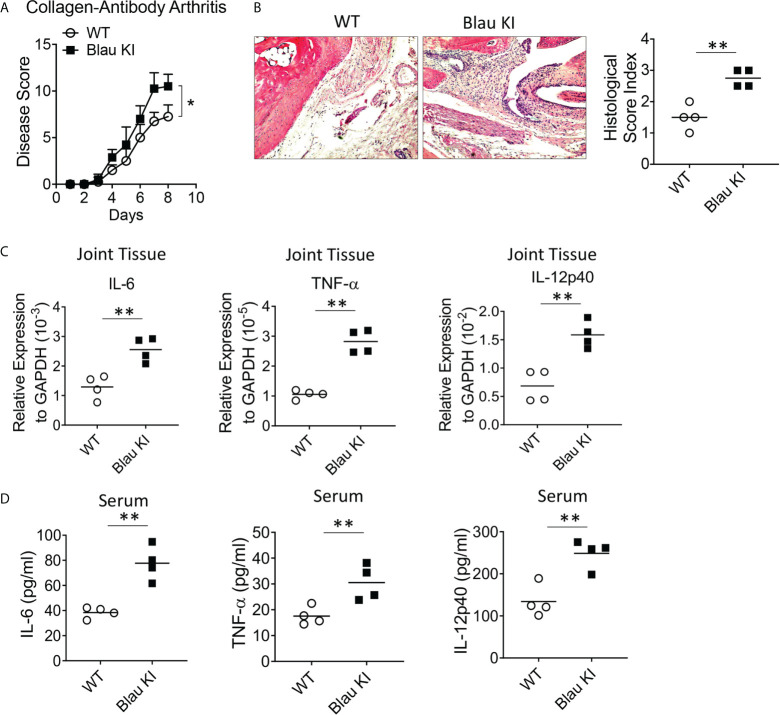
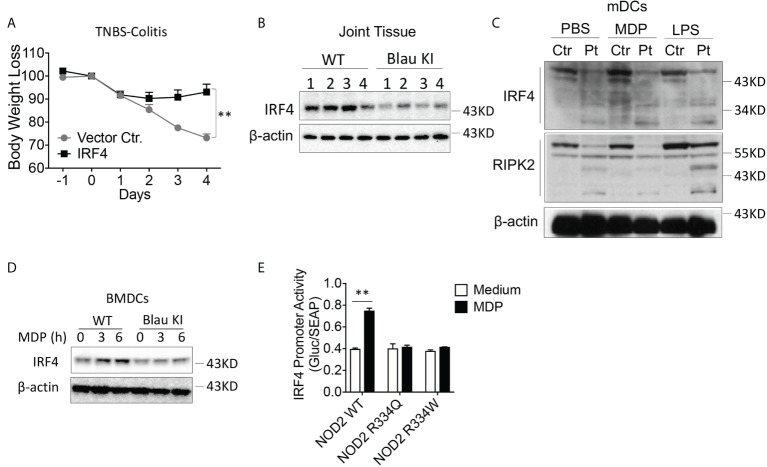
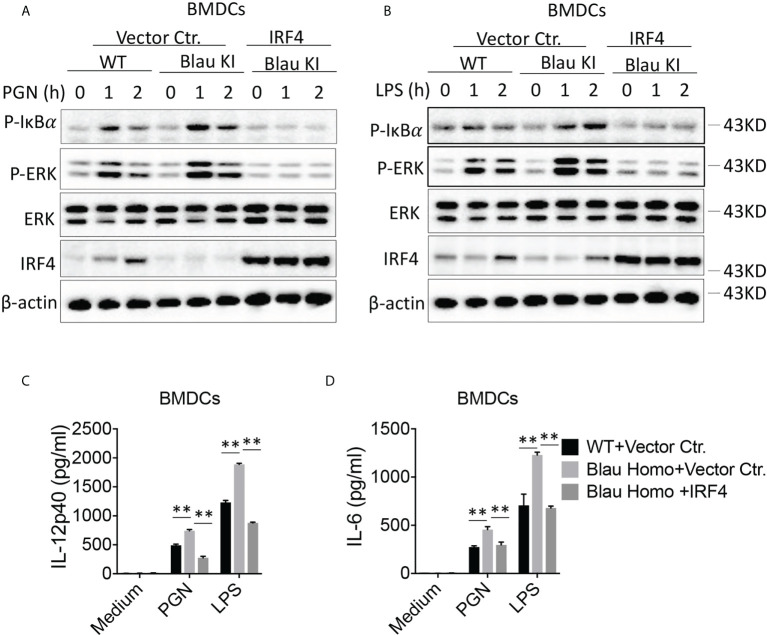
The studies described here provide an analysis of the pathogenesis of Blau syndrome and thereby the function of NOD2 as seen through the lens of its dysfunction resulting from Blau-associated NOD2 mutations in its nucleotide-binding domain (NBD). As such, this analysis also sheds light on the role of NOD2 risk polymorphisms in the LRR domain occurring in Crohn's disease. The main finding was that Blau NOD2 mutations precipitate a loss of canonical NOD2 signaling via RIPK2 and that this loss has two consequences: first, it results in defective NOD2 ligand (MDP)-mediated NF-κB activation and second, it disrupts NOD2-mediated cross-regulation whereby NOD2 downregulates concomitant innate (TLR) responses. Strong evidence is also presented favoring the view that NOD2-mediated cross-regulation is under mechanistic control by IRF4 and that failure to up-regulate this factor because of faulty NOD2 signaling is the proximal cause of defective cross-regulation and the latter's effect on Blau syndrome inflammation. Overall, these studies highlight the role of NOD2 as a regulatory factor and thus provide additional insight into its function in inflammatory disease. Mutations in the nucleotide binding domain of the CARD15 (NOD2) gene underlie the granulomatous inflammation characterizing Blau syndrome (BS). In studies probing the mechanism of this inflammation we show here that NOD2 plasmids expressing various Blau mutations in HEK293 cells result in reduced NOD2 activation of RIPK2 and correspondingly reduced NOD2 activation of NF-κB. These in vitro studies of NOD2 signaling were accompanied by in vivo studies showing that BS-NOD2 also exhibit defects in cross-regulation of innate responses underlying inflammation. Thus, whereas over-expressed intact NOD2 suppresses TNBS-colitis, over-expressed BS-NOD2 does not; in addition, whereas administration of NOD2 ligand (muramyl dipeptide, MDP) suppresses DSS-colitis in Wild Type (WT) mice it fails to do so in homozygous or heterozygous mice bearing a NOD2 Blau mutation. Similarly, mice bearing a Blau mutation exhibit enhanced anti-collagen antibody-induced arthritis. The basis of such cross-regulatory failure was revealed in studies showing that MDP-stimulated cells bearing BS-NOD2 exhibit a reduced capacity to signal via RIPK2 as well as a reduced capacity to up-regulate IRF4, a factor shown previously to mediate NOD2 suppression of NF-κB activation. Indeed, TLR-stimulated cells bearing a Blau mutation exhibited enhanced in vitro cytokine responses that are quieted by lentivirus transduction of IRF4. In addition, enhanced anti-collagen-induced joint inflammation in mice bearing a Blau mutation was accompanied by reduced IRF4 expression in inflamed joint tissue and IRF4 expression was reduced in MDP-stimulated cells from BS patients. Thus, inflammation characterizing Blau syndrome are caused, at least in part, by faulty canonical signaling and reduce IRF4-mediated cross-regulation.
Keywords: Blau; Crohns disease; IRF4; NFkapapB; Nod2.
Copyright © 2022 Mao, Dhar, Meng, Fuss, Montgomery-Recht, Yang, Xu, Kitani and Strober.
Conflict of interest statement
Author KM-R is employed by Leidos Biomedical Research, Inc. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
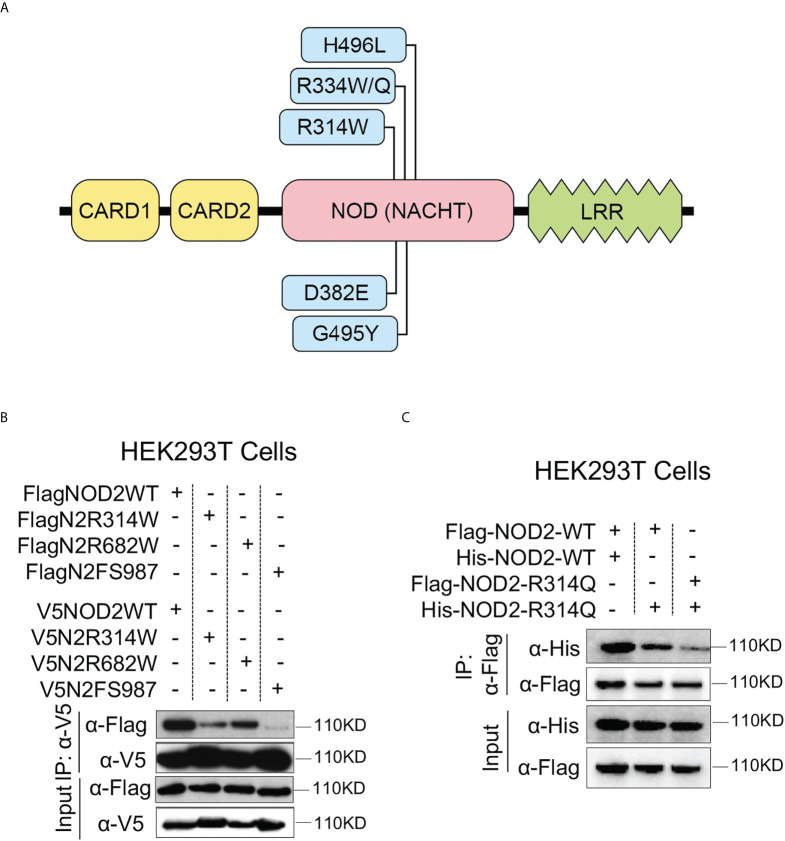
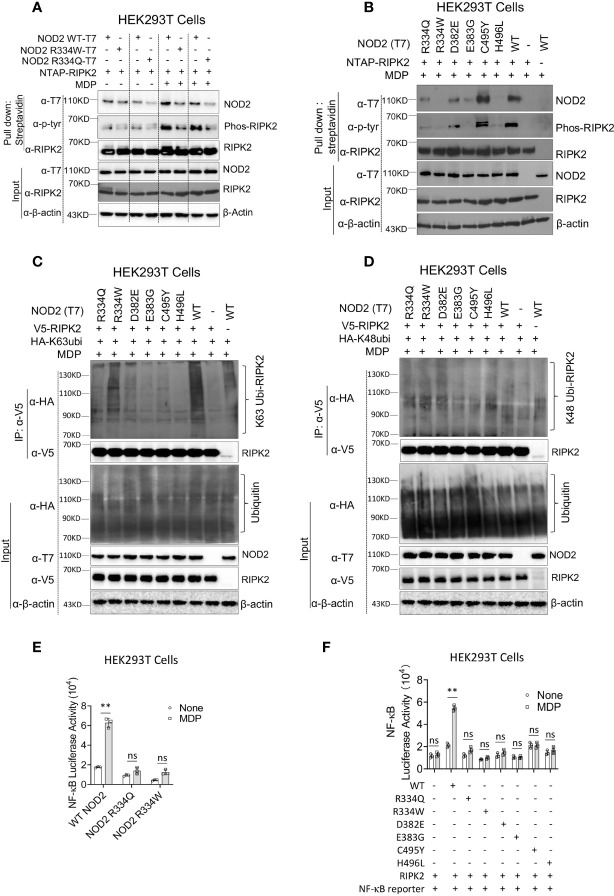
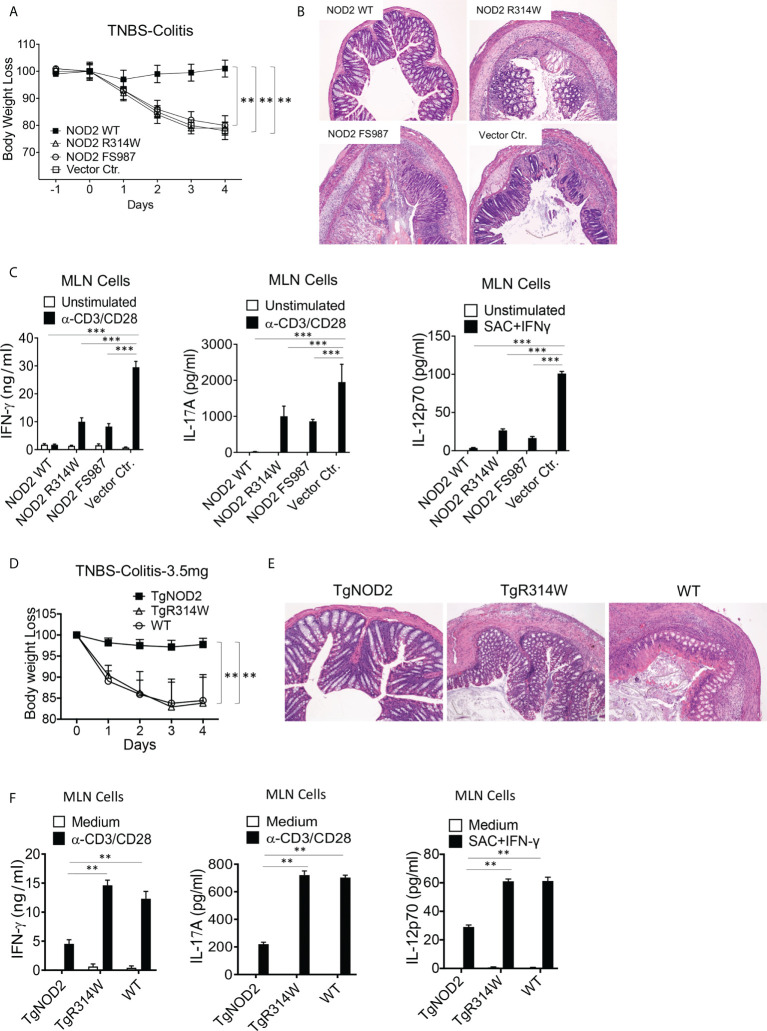
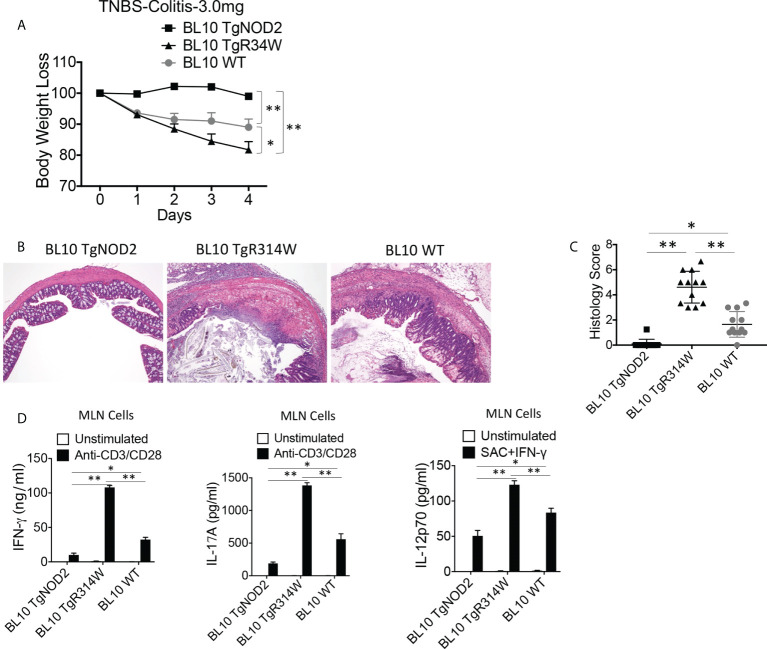
References
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials