

Blockades of effector T cell senescence and exhaustion synergistically enhance antitumor immunity and immunotherapy
- PMID: 36192086
- PMCID: PMC9535198
- DOI: 10.1136/jitc-2022-005020
Blockades of effector T cell senescence and exhaustion synergistically enhance antitumor immunity and immunotherapy
Abstract
Background: Current immunotherapies still have limited successful rates among cancers. It is now recognized that T cell functional state in the tumor microenvironment (TME) is a key determinant for effective antitumor immunity and immunotherapy. In addition to exhaustion, cellular senescence in tumor-infiltrating T cells (TILs) has recently been identified as an important T cell dysfunctional state induced by various malignant tumors. Therefore, a better understanding of the molecular mechanism responsible for T cell senescence in the TME and development of novel strategies to prevent effector T cell senescence are urgently needed for cancer immunotherapy.
Methods: Senescent T cell populations in the TMEs in mouse lung cancer, breast cancer, and melanoma tumor models were evaluated. Furthermore, T cell senescence induced by mouse tumor and regulatory T (Treg) cells in vitro was determined with multiple markers and assays, including real-time PCR, flow cytometry, and histochemistry staining. Loss-of-function strategies with pharmacological inhibitors and the knockout mouse model were used to identify the potential molecules and pathways involved in T cell senescence. In addition, melanoma mouse tumor immunotherapy models were performed to explore the synergistical efficacy of antitumor immunity via prevention of tumor-specific T cell senescence combined with anti-programmed death-ligand 1 (anti-PD-L1) checkpoint blockade therapy.
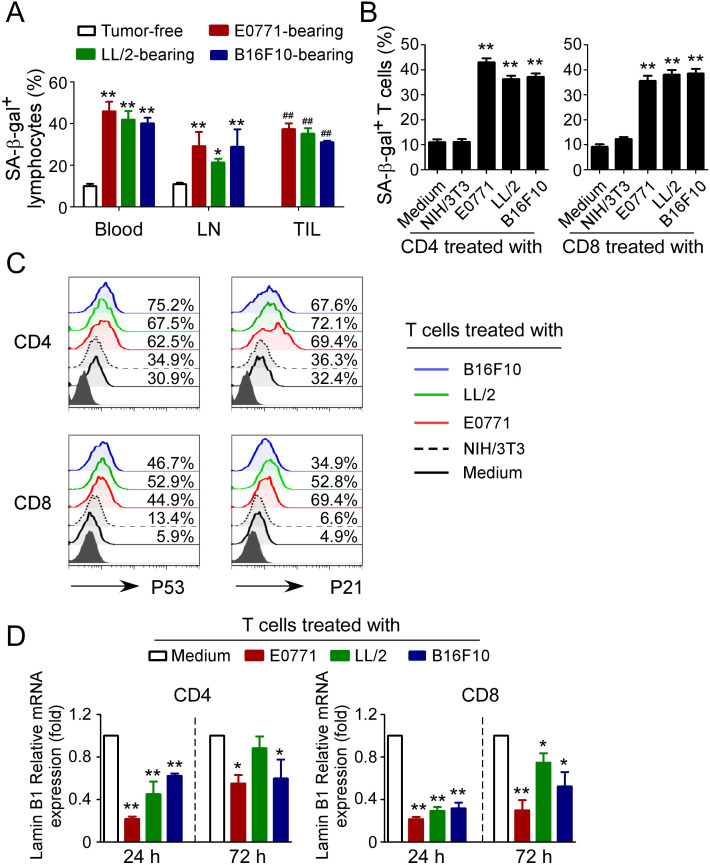
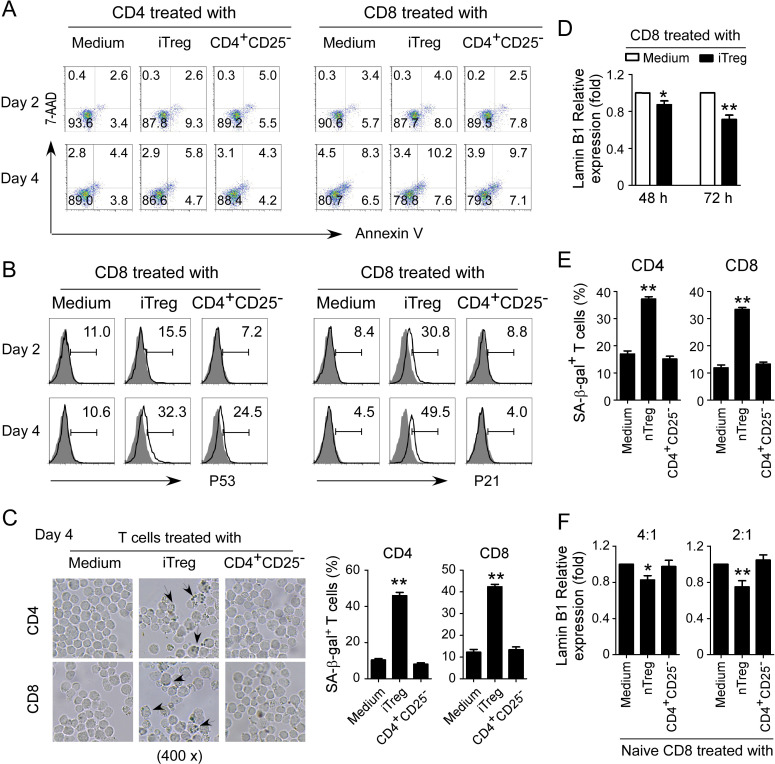
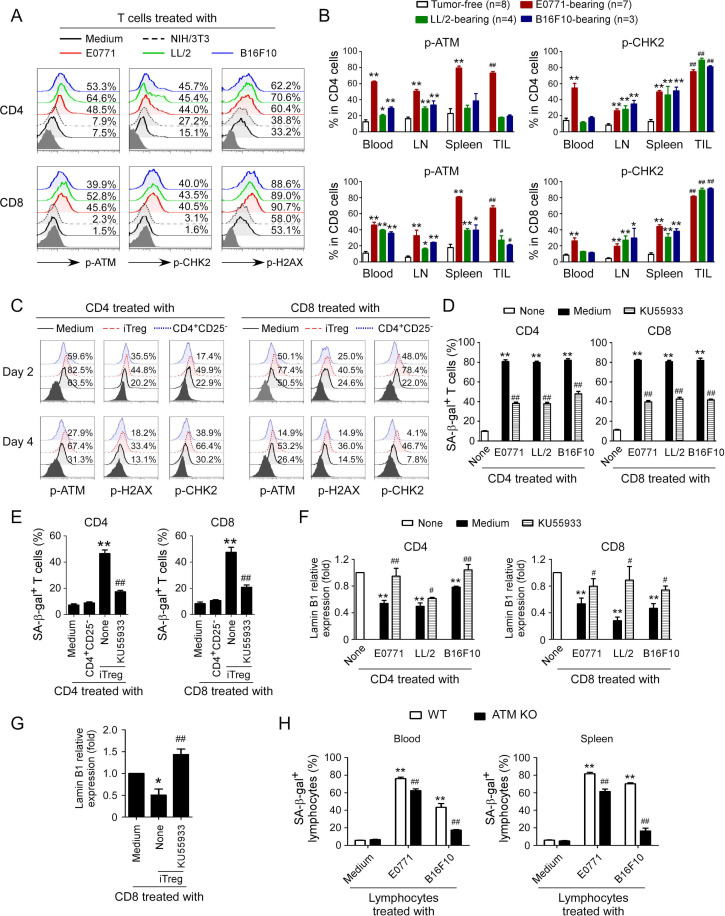
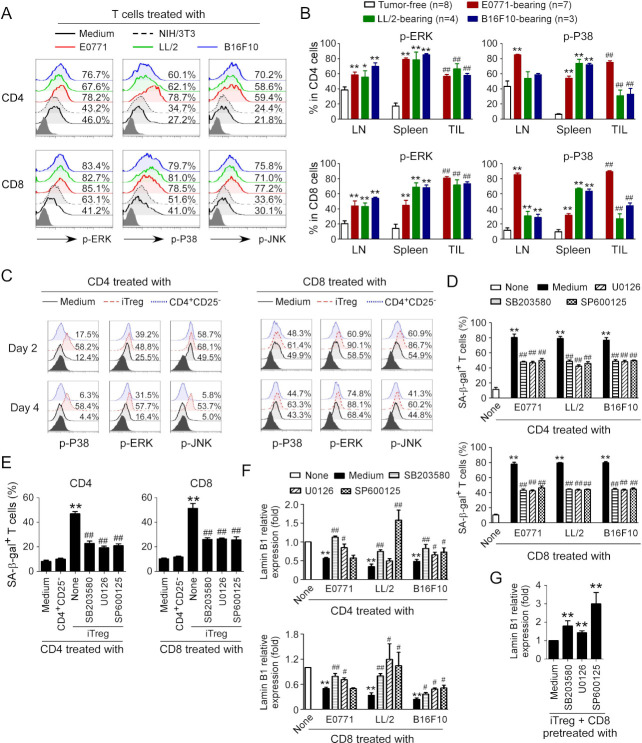
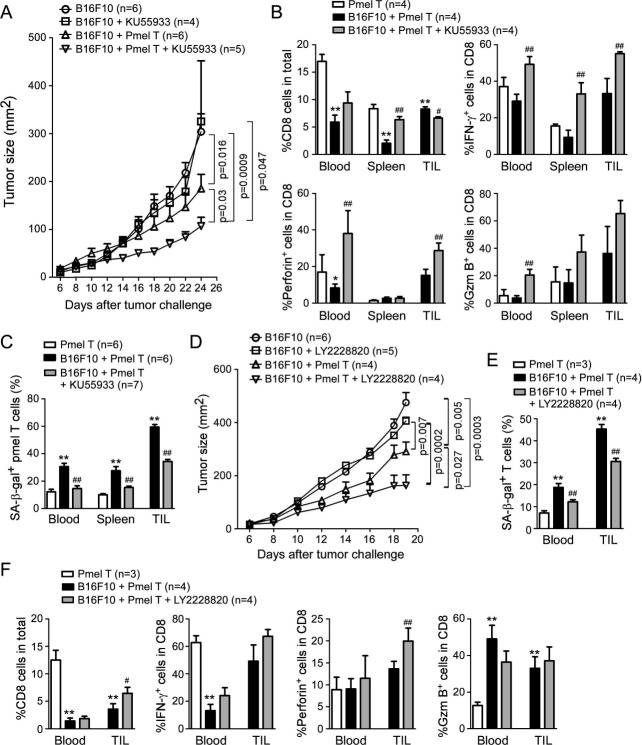
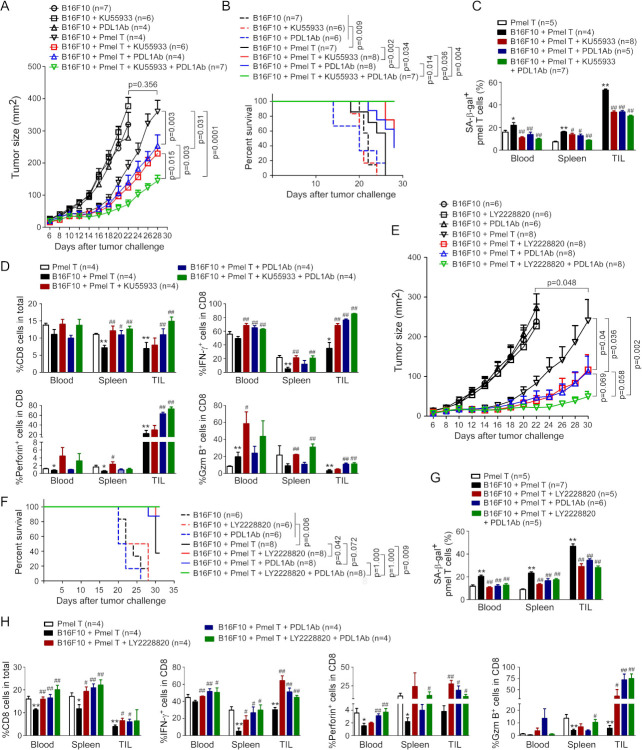
Results: We report that both mouse malignant tumor cells and Treg cells can induce responder T cell senescence, similar as shown in human Treg and tumor cells. Accumulated senescent T cells also exist in the TME in tumor models of lung cancer, breast cancer and melanoma. Induction of ataxia-telangiectasia mutated protein (ATM)-associated DNA damage is the cause for T cell senescence induced by both mouse tumor cells and Treg cells, which is also regulated by mitogen-activated protein kinase (MAPK) signaling. Furthermore, blockages of ATM-associated DNA damage and/or MAPK signaling pathways in T cells can prevent T cell senescence mediated by tumor cells and Treg cells in vitro and enhance antitumor immunity and immunotherapy in vivo in adoptive transfer T cell therapy melanoma models. Importantly, prevention of tumor-specific T cell senescence via ATM and/or MAPK signaling inhibition combined with anti-PD-L1 checkpoint blockade can synergistically enhance antitumor immunity and immunotherapy in vivo.
Conclusions: These studies prove the novel concept that targeting both effector T cell senescence and exhaustion is an effective strategy and can synergistically enhance cancer immunotherapy.
Keywords: Immunotherapy; Melanoma; T-Lymphocytes; Tumor Escape; Tumor Microenvironment.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
Similar articles
-
Blocking senescence and tolerogenic function of dendritic cells induced by γδ Treg cells enhances tumor-specific immunity for cancer immunotherapy.J Immunother Cancer. 2024 Apr 5;12(4):e008219. doi: 10.1136/jitc-2023-008219. J Immunother Cancer. 2024. PMID: 38580332 Free PMC article.
-
Tumor-derived ILT4 induces T cell senescence and suppresses tumor immunity.J Immunother Cancer. 2021 Mar;9(3):e001536. doi: 10.1136/jitc-2020-001536. J Immunother Cancer. 2021. PMID: 33653799 Free PMC article.
-
Future perspectives in melanoma research : Meeting report from the "Melanoma Bridge". Napoli, December 1st-4th 2015.J Transl Med. 2016 Nov 15;14(1):313. doi: 10.1186/s12967-016-1070-y. J Transl Med. 2016. PMID: 27846884 Free PMC article.
-
Exhaustion and senescence: two crucial dysfunctional states of T cells in the tumor microenvironment.Cell Mol Immunol. 2020 Jan;17(1):27-35. doi: 10.1038/s41423-019-0344-8. Epub 2019 Dec 18. Cell Mol Immunol. 2020. PMID: 31853000 Free PMC article. Review.
-
T cell exhaustion and senescence for ovarian cancer immunotherapy.Semin Cancer Biol. 2024 Sep;104-105:1-15. doi: 10.1016/j.semcancer.2024.07.001. Epub 2024 Jul 18. Semin Cancer Biol. 2024. PMID: 39032717 Review.
Cited by
-
The Cross Talk between Cellular Senescence and Melanoma: From Molecular Pathogenesis to Target Therapies.Cancers (Basel). 2023 May 6;15(9):2640. doi: 10.3390/cancers15092640. Cancers (Basel). 2023. PMID: 37174106 Free PMC article. Review.
-
Chemoradiotherapy-induced ACKR2+ tumor cells drive CD8+ T cell senescence and cervical cancer recurrence.Cell Rep Med. 2024 May 21;5(5):101550. doi: 10.1016/j.xcrm.2024.101550. Epub 2024 May 8. Cell Rep Med. 2024. PMID: 38723624 Free PMC article.
-
Lipid metabolic reprograming: the unsung hero in breast cancer progression and tumor microenvironment.Mol Cancer. 2025 Mar 3;24(1):61. doi: 10.1186/s12943-025-02258-1. Mol Cancer. 2025. PMID: 40025508 Free PMC article. Review.
-
Immunotherapeutic strategies in head and neck cancer: challenges and opportunities.J Clin Invest. 2025 Apr 15;135(8):e188128. doi: 10.1172/JCI188128. eCollection 2025 Apr 15. J Clin Invest. 2025. PMID: 40231472 Free PMC article. Review.
-
ATM Inhibition-Induced ISG15/IFI27/OASL Is Correlated with Immunotherapy Response and Inflamed Immunophenotype.Cells. 2023 Apr 30;12(9):1288. doi: 10.3390/cells12091288. Cells. 2023. PMID: 37174688 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous