

Ribosome-bound Upf1 forms distinct 80S complexes and conducts mRNA surveillance
- PMID: 36192133
- PMCID: PMC9670811
- DOI: 10.1261/rna.079416.122
Ribosome-bound Upf1 forms distinct 80S complexes and conducts mRNA surveillance
Abstract
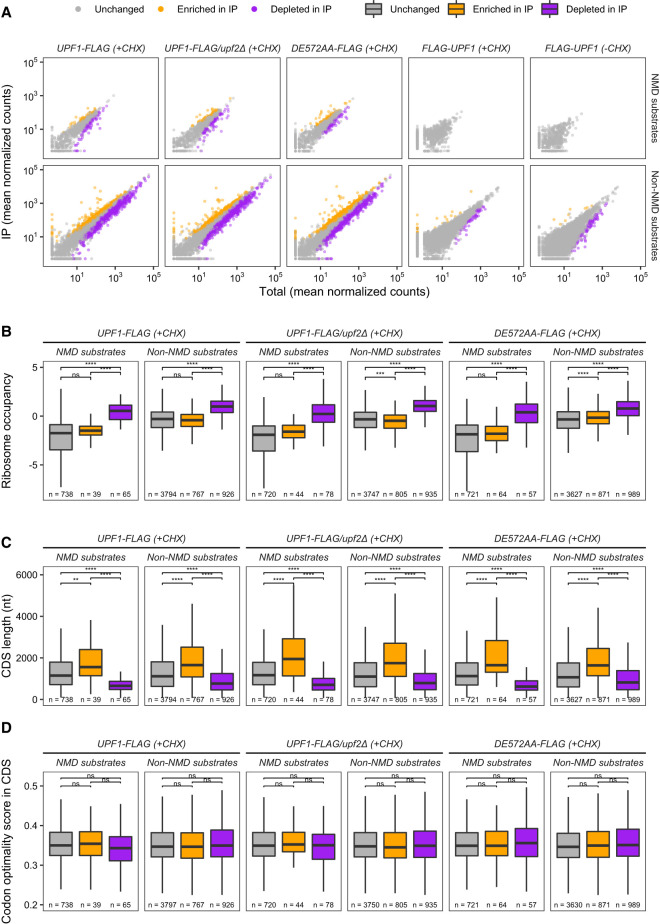
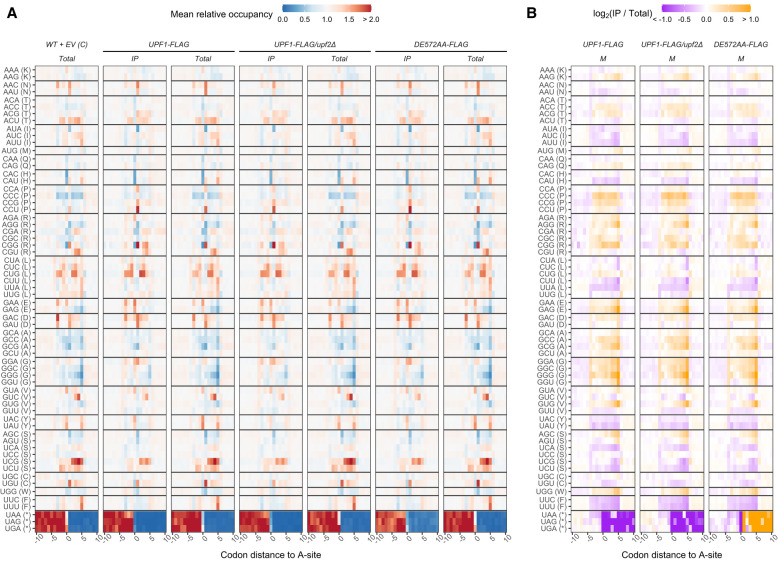
Upf1, Upf2, and Upf3, the central regulators of nonsense-mediated mRNA decay (NMD), appear to exercise their NMD functions while bound to elongating ribosomes, and evidence for this conclusion is particularly compelling for Upf1. Hence, we used selective profiling of yeast Upf1:ribosome association to define that step in greater detail, understand whether the nature of the mRNA being translated influences Upf1:80S interaction, and elucidate the functions of ribosome-associated Upf1. Our approach has allowed us to clarify the timing and specificity of Upf1 association with translating ribosomes, obtain evidence for a Upf1 mRNA surveillance function that precedes the activation of NMD, identify a unique ribosome state that generates 37-43 nt ribosome footprints whose accumulation is dependent on Upf1's ATPase activity, and demonstrate that a mutated form of Upf1 can interfere with normal translation termination and ribosome release. In addition, our results strongly support the existence of at least two distinct functional Upf1 complexes in the NMD pathway.
Keywords: NMD substrates; Upf proteins; selective ribosome profiling.
© 2022 Ganesan et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures







Similar articles
-
Inhibition of post-termination ribosome recycling at premature termination codons in UPF1 ATPase mutants.Elife. 2020 Jul 22;9:e57834. doi: 10.7554/eLife.57834. Elife. 2020. PMID: 32697194 Free PMC article.
-
Genetic screens in Saccharomyces cerevisiae identify a role for 40S ribosome recycling factors Tma20 and Tma22 in nonsense-mediated decay.G3 (Bethesda). 2024 Mar 6;14(3):jkad295. doi: 10.1093/g3journal/jkad295. G3 (Bethesda). 2024. PMID: 38198768 Free PMC article.
-
Selective profiling of ribosomes associated with yeast Upf proteins.Methods. 2019 Feb 15;155:58-67. doi: 10.1016/j.ymeth.2018.12.008. Epub 2018 Dec 26. Methods. 2019. PMID: 30593864 Free PMC article.
-
NMD monitors translational fidelity 24/7.Curr Genet. 2017 Dec;63(6):1007-1010. doi: 10.1007/s00294-017-0709-4. Epub 2017 May 23. Curr Genet. 2017. PMID: 28536849 Free PMC article. Review.
-
New insights into the interplay between the translation machinery and nonsense-mediated mRNA decay factors.Biochem Soc Trans. 2018 Jun 19;46(3):503-512. doi: 10.1042/BST20170427. Epub 2018 Apr 6. Biochem Soc Trans. 2018. PMID: 29626148 Free PMC article. Review.
Cited by
-
Sensing of H2O2-induced oxidative stress by the UPF factor complex is crucial for activation of catalase-3 expression in Neurospora.PLoS Genet. 2023 Oct 16;19(10):e1010985. doi: 10.1371/journal.pgen.1010985. eCollection 2023 Oct. PLoS Genet. 2023. PMID: 37844074 Free PMC article.
-
Translation-coupled mRNA quality control mechanisms.EMBO J. 2023 Oct 4;42(19):e114378. doi: 10.15252/embj.2023114378. Epub 2023 Aug 22. EMBO J. 2023. PMID: 37605642 Free PMC article. Review.
-
RAPIDASH: A tag-free enrichment of ribosome-associated proteins reveals compositional dynamics in embryonic tissues and stimulated macrophages.bioRxiv [Preprint]. 2023 Dec 7:2023.12.07.570613. doi: 10.1101/2023.12.07.570613. bioRxiv. 2023. Update in: Mol Cell. 2024 Sep 19;84(18):3545-3563.e25. doi: 10.1016/j.molcel.2024.08.023. PMID: 38106052 Free PMC article. Updated. Preprint.
-
UPF1-From mRNA Degradation to Human Disorders.Cells. 2023 Jan 27;12(3):419. doi: 10.3390/cells12030419. Cells. 2023. PMID: 36766761 Free PMC article. Review.
-
Nonsense-Mediated mRNA Decay Factor Functions in Human Health and Disease.Biomedicines. 2023 Feb 27;11(3):722. doi: 10.3390/biomedicines11030722. Biomedicines. 2023. PMID: 36979701 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials