

The Phenotypic Continuum of ATP1A3-Related Disorders
- PMID: 36192182
- PMCID: PMC9576304
- DOI: 10.1212/WNL.0000000000200927
The Phenotypic Continuum of ATP1A3-Related Disorders
Abstract
Background and objectives: ATP1A3 is associated with a broad spectrum of predominantly neurologic disorders, which continues to expand beyond the initially defined phenotypes of alternating hemiplegia of childhood, rapid-onset dystonia parkinsonism, and cerebellar ataxia, areflexia, pes cavus, optic atrophy, sensorineural hearing loss syndrome. This phenotypic variability makes it challenging to assess the pathogenicity of an ATP1A3 variant found in an undiagnosed patient. We describe the phenotypic features of individuals carrying a pathogenic/likely pathogenic ATP1A3 variant and perform a literature review of all ATP1A3 variants published thus far in association with human neurologic disease. Our aim is to demonstrate the heterogeneous clinical spectrum of the gene and look for phenotypic overlap between patients that will streamline the diagnostic process.
Methods: Undiagnosed individuals with ATP1A3 variants were identified within the cohort of the Deciphering Developmental Disorders study with additional cases contributed by collaborators internationally. Detailed clinical data were collected with consent through a questionnaire completed by the referring clinicians. PubMed was searched for publications containing the term "ATP1A3" from 2004 to 2021.
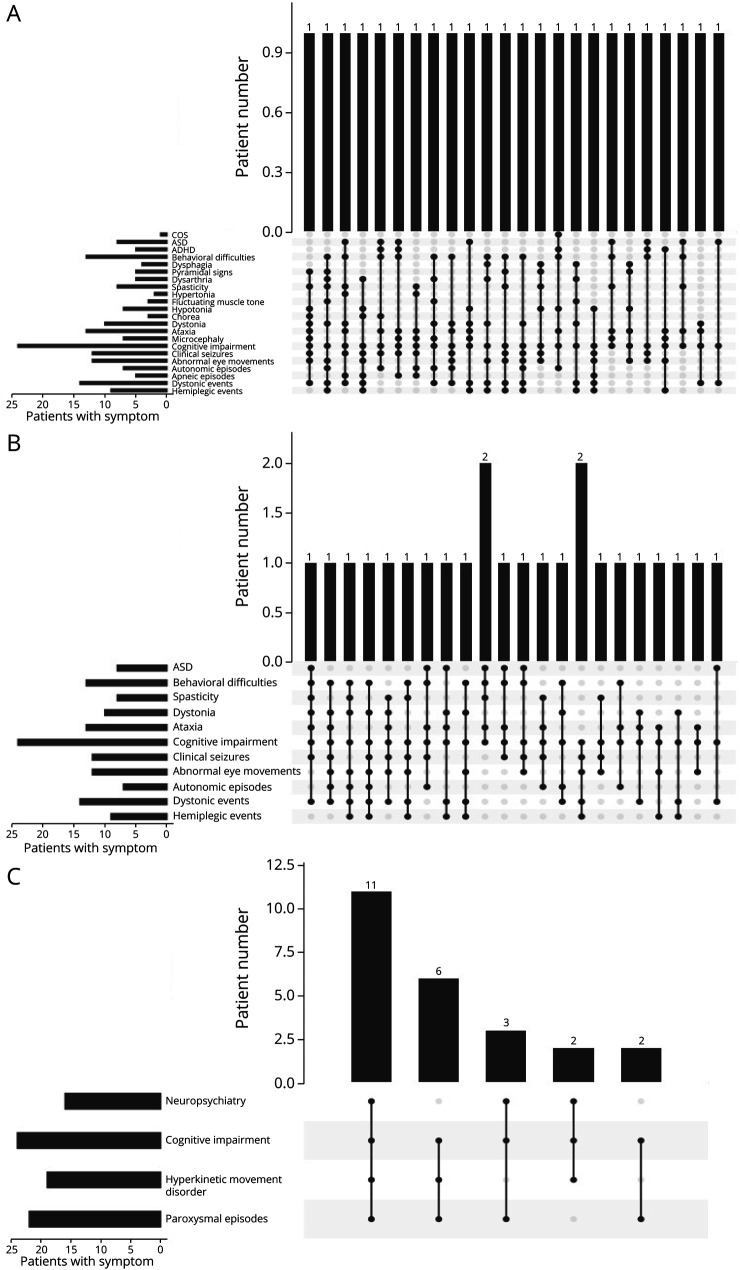
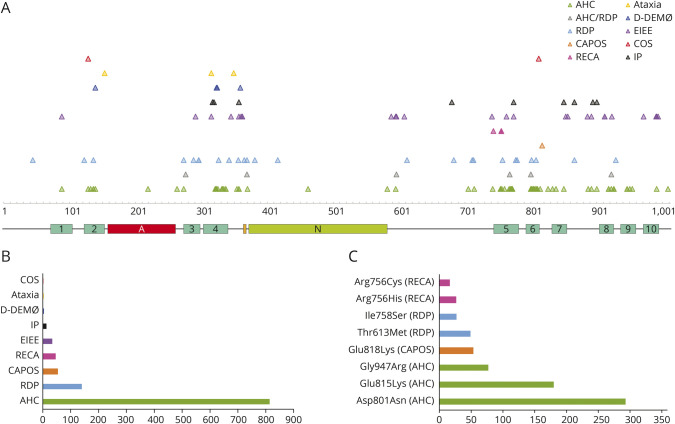
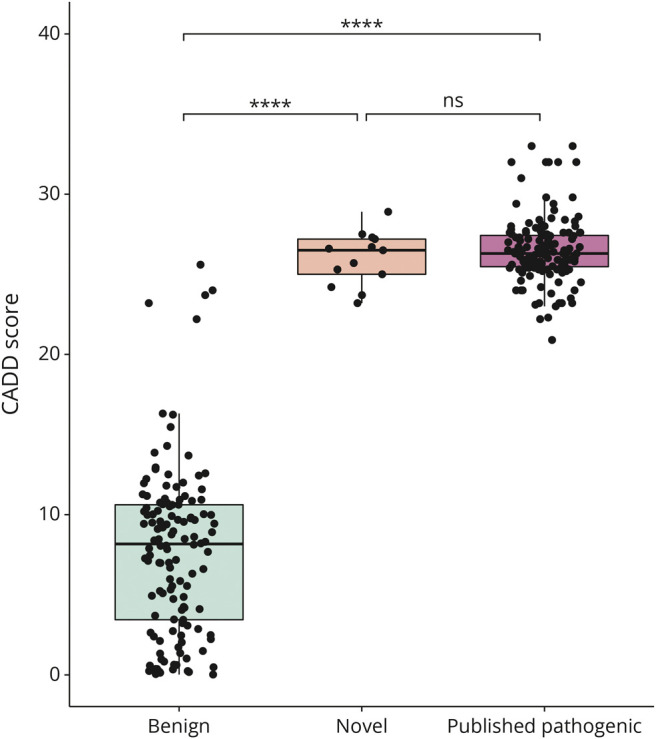
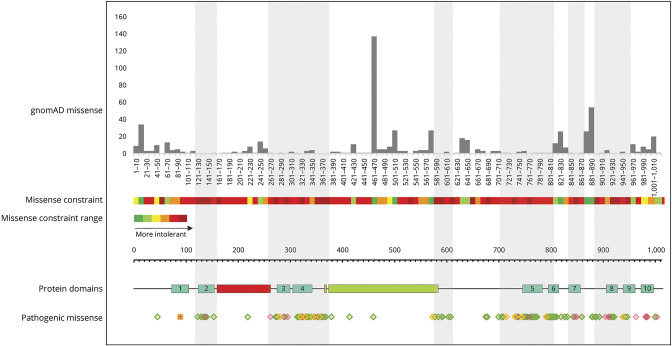
Results: Twenty-four individuals with a previously undiagnosed neurologic phenotype were found to carry 21 ATP1A3 variants. Eight variants have been previously published. Patients experienced on average 2-3 different types of paroxysmal events. Permanent neurologic features were common including microcephaly (7; 29%), ataxia (13; 54%), dystonia (10; 42%), and hypotonia (7; 29%). All patients had cognitive impairment. Neuropsychiatric diagnoses were reported in 16 (66.6%) individuals. Phenotypes were extremely varied, and most individuals did not fit clinical criteria for previously published phenotypes. On review of the literature, 1,108 individuals have been reported carrying 168 different ATP1A3 variants. The most common variants are associated with well-defined phenotypes, while more rare variants often result in very rare symptom correlations, such as are seen in our study. Combined Annotation-Dependent Depletion (CADD) scores of pathogenic and likely pathogenic variants were significantly higher and variants clustered within 6 regions of constraint.
Discussion: Our study shows that looking for a combination of paroxysmal events, hyperkinesia, neuropsychiatric symptoms, and cognitive impairment and evaluating the CADD score and variant location can help identify an ATP1A3-related condition, rather than applying diagnostic criteria alone.
Copyright © 2022 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources