

HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma
- PMID: 36192599
- PMCID: PMC9584821
- DOI: 10.1038/s42255-022-00642-5
HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma
Abstract
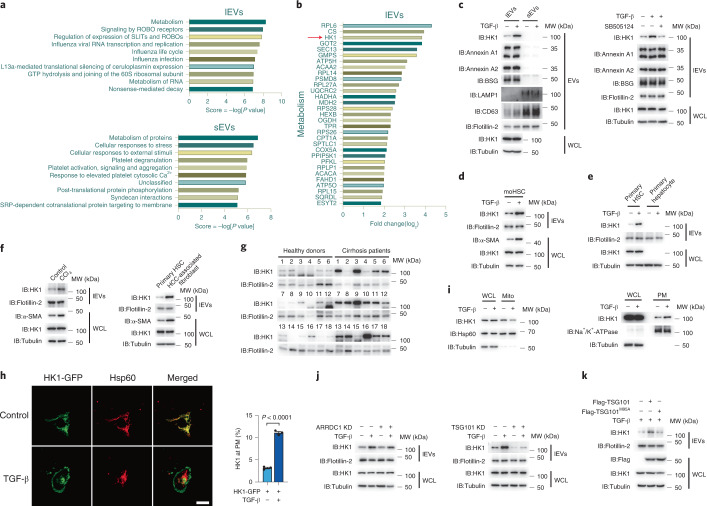
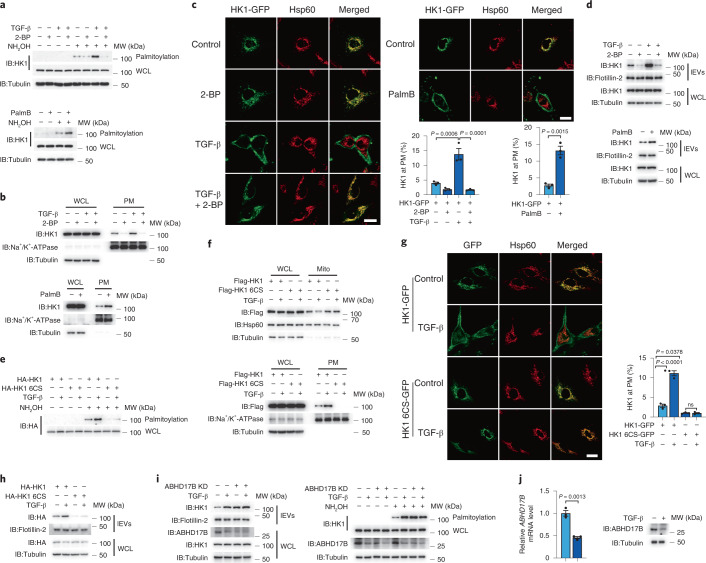
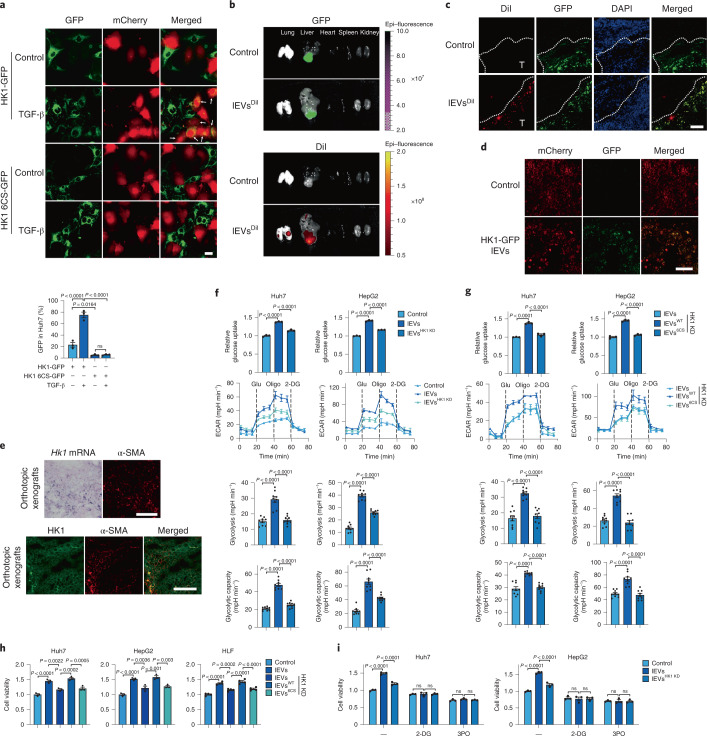
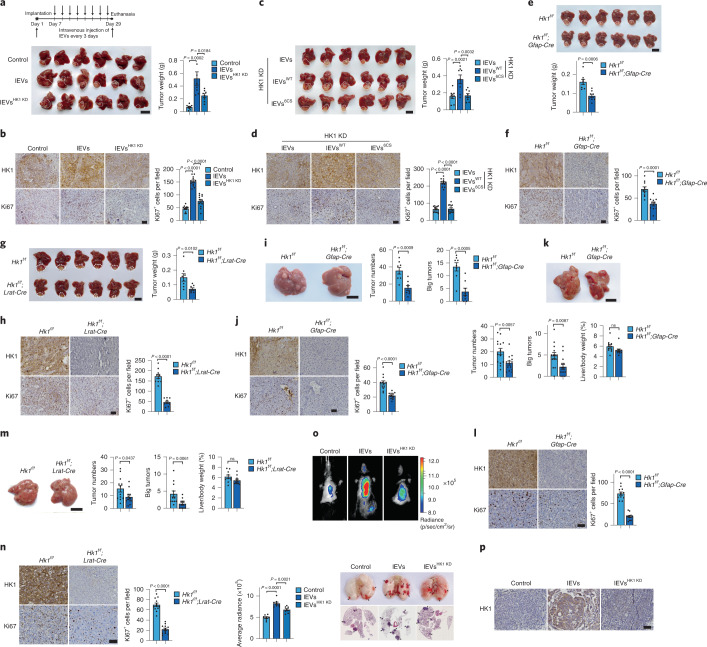
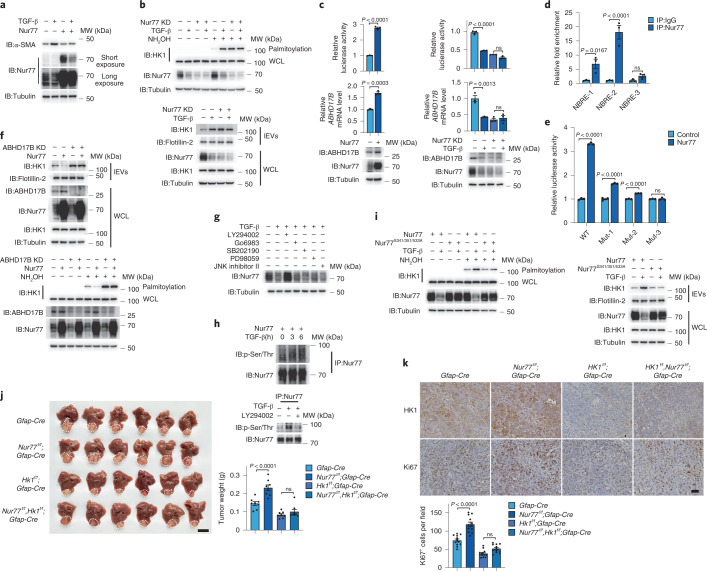
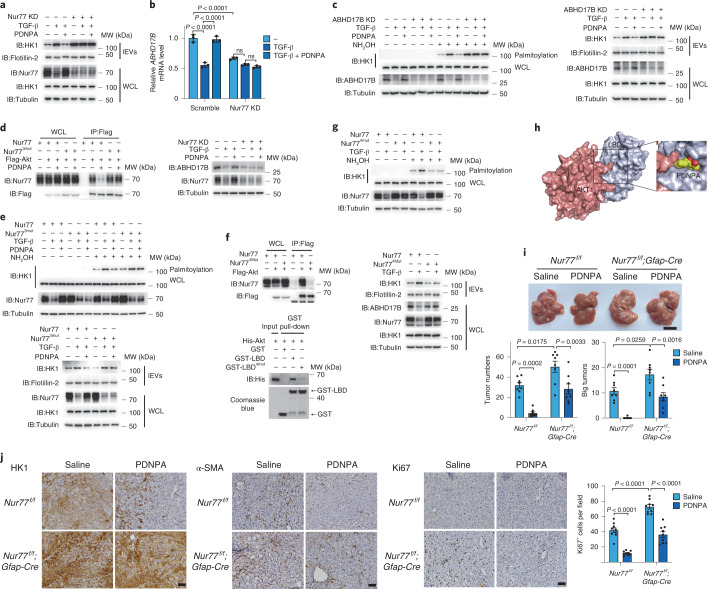
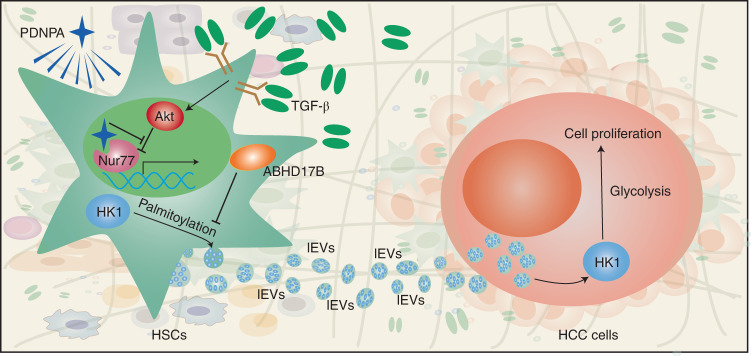
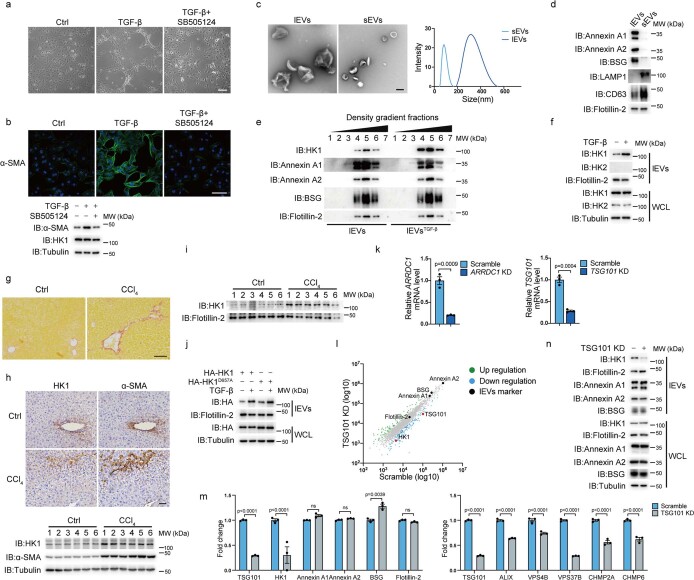
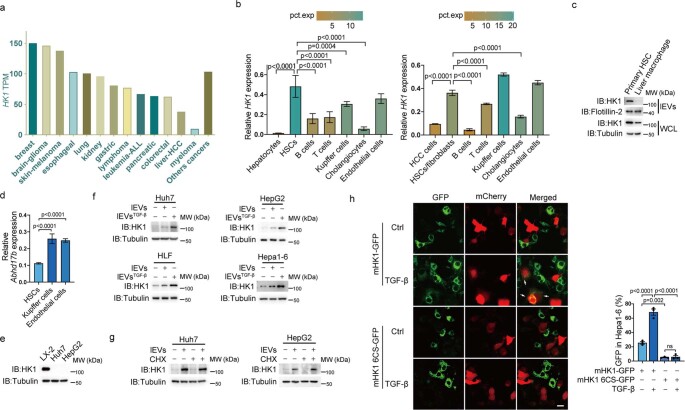
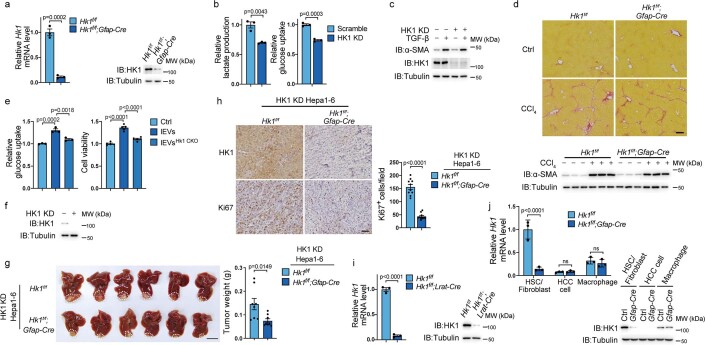
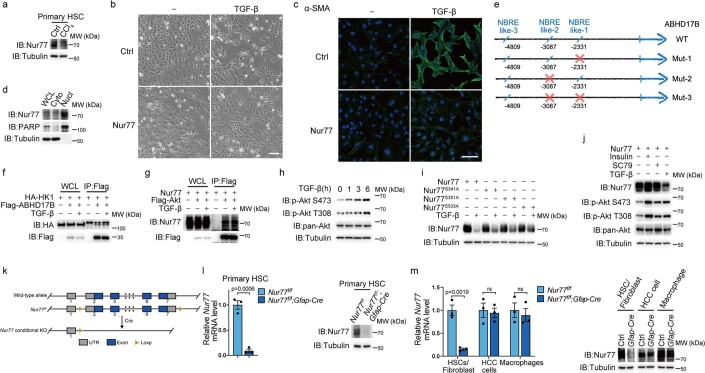
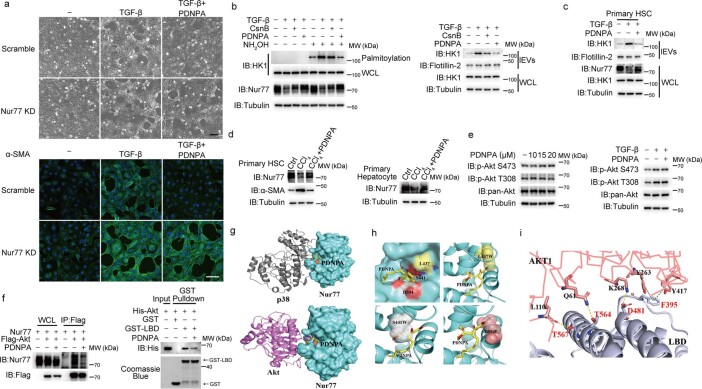
Extracellular vesicles play crucial roles in intercellular communication in the tumor microenvironment. Here we demonstrate that in hepatic fibrosis, TGF-β stimulates the palmitoylation of hexokinase 1 (HK1) in hepatic stellate cells (HSCs), which facilitates the secretion of HK1 via large extracellular vesicles in a TSG101-dependent manner. The large extracellular vesicle HK1 is hijacked by hepatocellular carcinoma (HCC) cells, leading to accelerated glycolysis and HCC progression. In HSCs, the nuclear receptor Nur77 transcriptionally activates the expression of depalmitoylase ABHD17B to inhibit HK1 palmitoylation, consequently attenuating HK1 release. However, TGF-β-activated Akt functionally represses Nur77 by inducing Nur77 phosphorylation and degradation. We identify the small molecule PDNPA that binds Nur77 to generate steric hindrance to block Akt targeting, thereby disrupting Akt-mediated Nur77 degradation and preserving Nur77 inhibition of HK1 release. Together, this study demonstrates an overlooked function of HK1 in HCC upon its release from HSCs and highlights PDNPA as a candidate compound for inhibiting HCC progression.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
Liver cancer metabolism: a hexokinase from the stars.Nat Metab. 2022 Oct;4(10):1225-1226. doi: 10.1038/s42255-022-00659-w. Nat Metab. 2022. PMID: 36192600 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
