

Epidermal Growth Factor Receptor Inhibition Is Protective in Hyperoxia-Induced Lung Injury
- PMID: 36193076
- PMCID: PMC9526641
- DOI: 10.1155/2022/9518592
Epidermal Growth Factor Receptor Inhibition Is Protective in Hyperoxia-Induced Lung Injury
Abstract
Aims: Studies have linked severe hyperoxia, or prolonged exposure to very high oxygen levels, with worse clinical outcomes. This study investigated the role of epidermal growth factor receptor (EGFR) in hyperoxia-induced lung injury at very high oxygen levels (>95%).
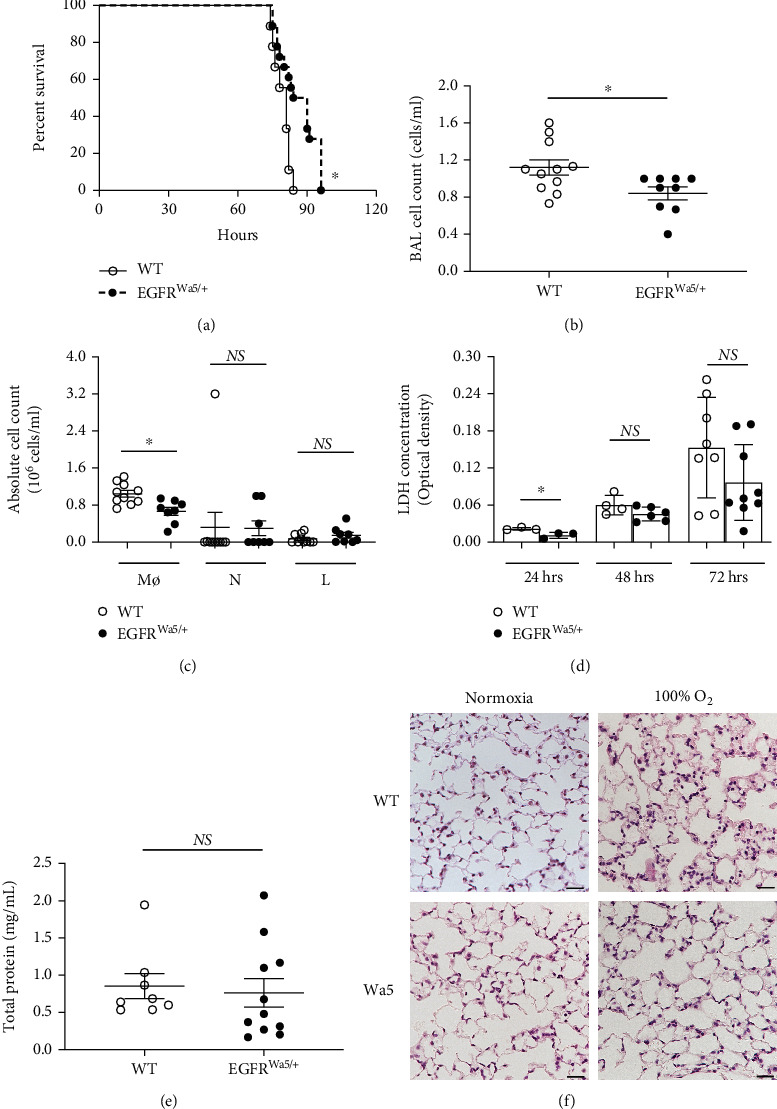
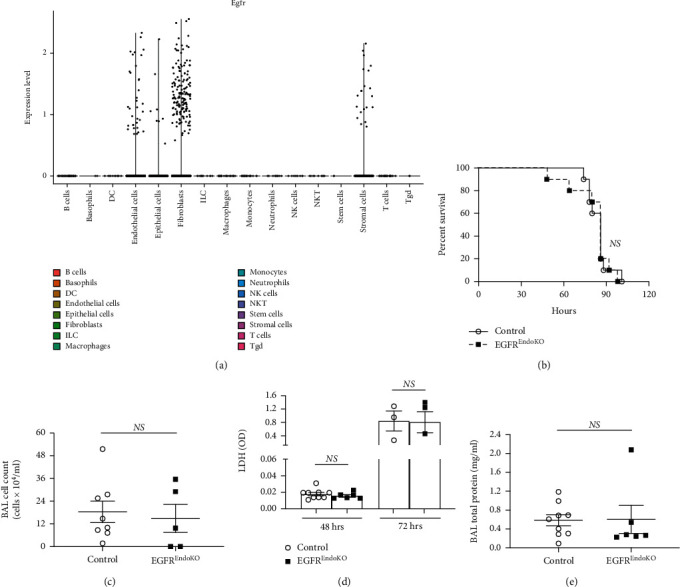
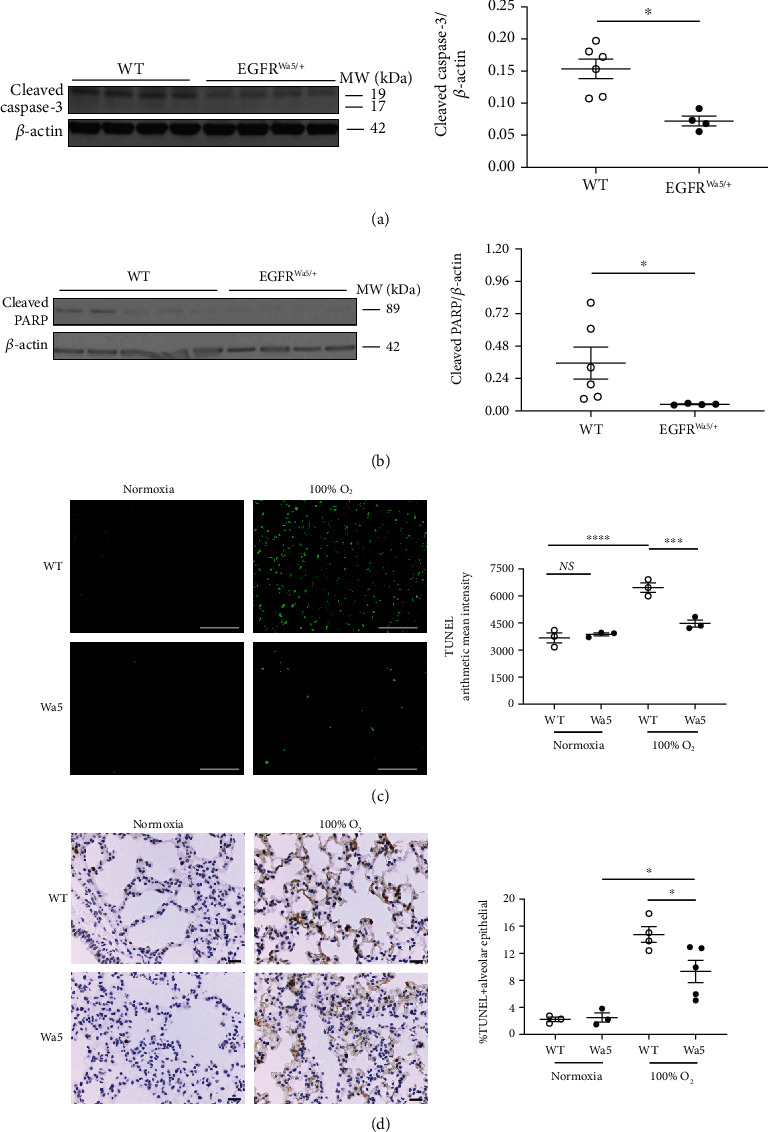
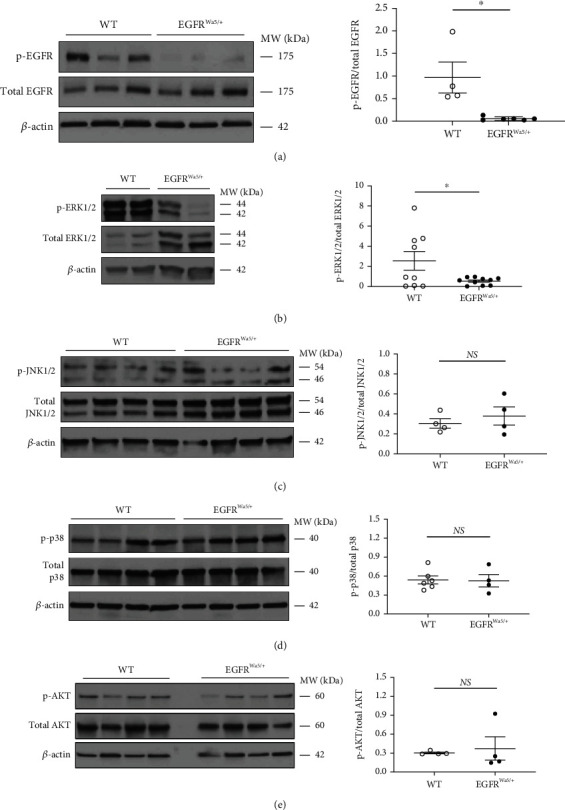
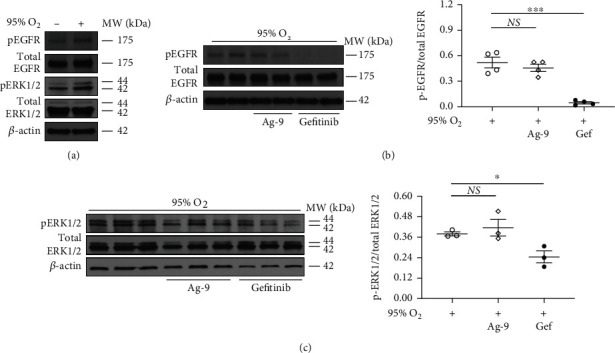
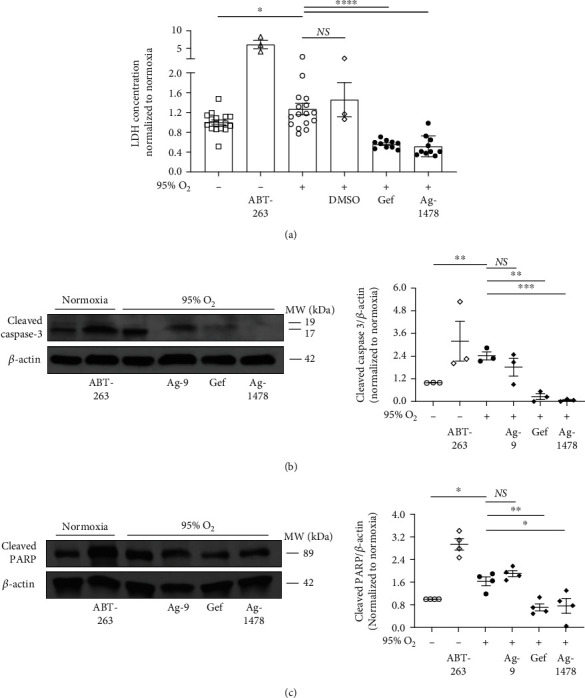
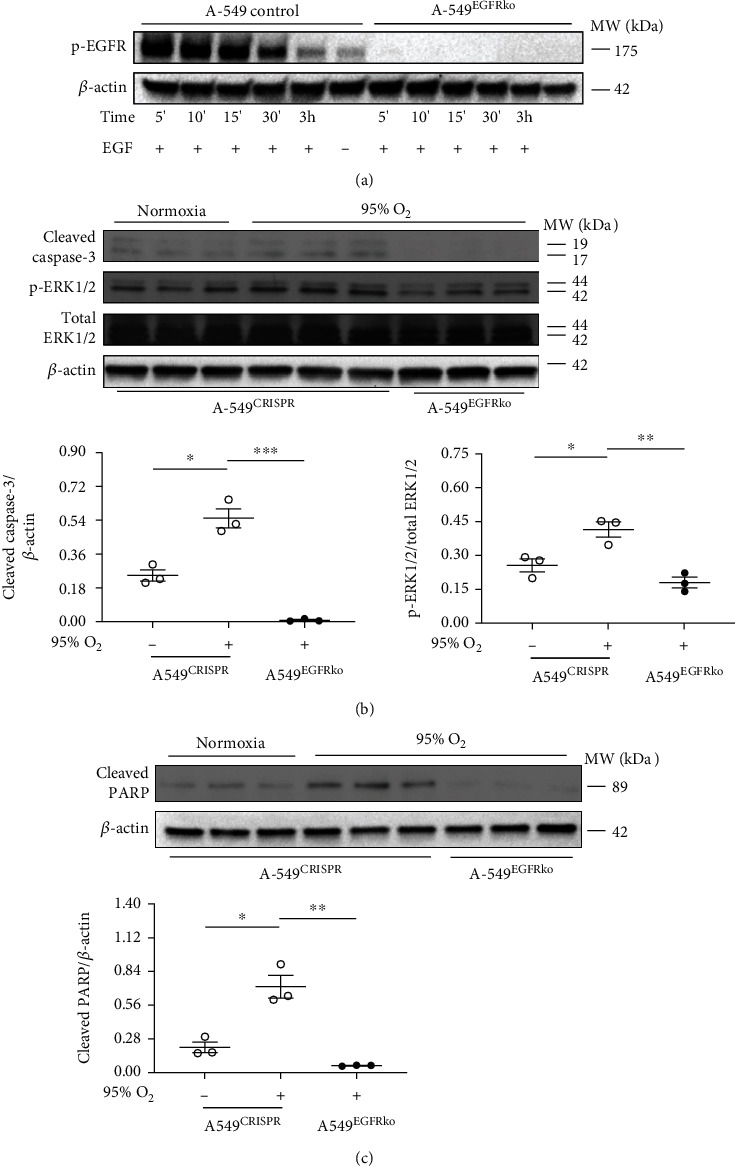
Results: Effects of severe hyperoxia (100% oxygen) were studied in mice with genetically inhibited EGFR and wild-type littermates. Despite the established role of EGFR in lung repair, EGFR inhibition led to improved survival and reduced acute lung injury, which prompted an investigation into this protective mechanism. Endothelial EGFR genetic knockout did not confer protection. EGFR inhibition led to decreased levels of cleaved caspase-3 and poly (ADP-ribosyl) polymerase (PARP) and decreased terminal dUTP nick end labeling- (TUNEL-) positive staining in alveolar epithelial cells and reduced ERK activation, which suggested reduced apoptosis in vivo. EGFR inhibition decreased hyperoxia (95%)-induced apoptosis and ERK in murine alveolar epithelial cells in vitro, and CRISPR-mediated EGFR deletion reduced hyperoxia-induced apoptosis and ERK in human alveolar epithelial cells in vitro. Innovation. This work defines a protective role of EGFR inhibition to decrease apoptosis in lung injury induced by 100% oxygen. This further characterizes the complex role of EGFR in acute lung injury and outlines a novel hyperoxia-induced cell death pathway that warrants further study.
Conclusion: In conditions of severe hyperoxia (>95% for >24 h), EGFR inhibition led to improved survival, decreased lung injury, and reduced cell death. These findings further elucidate the complex role of EGFR in acute lung injury.
Copyright © 2022 Zachary M. Harris et al.
Conflict of interest statement
The authors declare that there is no conflict of interest regarding the publication of this article.
Figures
Similar articles
-
Poly(ADP-ribose)polymerase activation mediates lung epithelial cell death in vitro but is not essential in hyperoxia-induced lung injury.Am J Respir Cell Mol Biol. 2005 Dec;33(6):555-64. doi: 10.1165/rcmb.2004-0361OC. Epub 2005 Sep 8. Am J Respir Cell Mol Biol. 2005. PMID: 16151053
-
Lung injury caused by high tidal volume mechanical ventilation and hyperoxia is dependent on oxidant-mediated c-Jun NH2-terminal kinase activation.J Appl Physiol (1985). 2011 Nov;111(5):1467-76. doi: 10.1152/japplphysiol.00539.2011. Epub 2011 Jul 28. J Appl Physiol (1985). 2011. PMID: 21799126 Free PMC article.
-
A paradoxical protective role for the proinflammatory peptide substance P receptor (NK1R) in acute hyperoxic lung injury.Am J Physiol Lung Cell Mol Physiol. 2009 Oct;297(4):L687-97. doi: 10.1152/ajplung.90509.2008. Epub 2009 Jul 24. Am J Physiol Lung Cell Mol Physiol. 2009. PMID: 19633070 Free PMC article.
-
Inhibition of Regulatory-Associated Protein of Mechanistic Target of Rapamycin Prevents Hyperoxia-Induced Lung Injury by Enhancing Autophagy and Reducing Apoptosis in Neonatal Mice.Am J Respir Cell Mol Biol. 2016 Nov;55(5):722-735. doi: 10.1165/rcmb.2015-0349OC. Am J Respir Cell Mol Biol. 2016. PMID: 27374190 Free PMC article.
-
Amphiregulin inhibits TNF-α-induced alveolar epithelial cell death through EGFR signaling pathway.Biomed Pharmacother. 2020 May;125:109995. doi: 10.1016/j.biopha.2020.109995. Epub 2020 Feb 27. Biomed Pharmacother. 2020. PMID: 32187954
Cited by
-
Optimized preparation pipeline for emergency phage therapy against Pseudomonas aeruginosa at Yale University.Sci Rep. 2024 Feb 1;14(1):2657. doi: 10.1038/s41598-024-52192-3. Sci Rep. 2024. PMID: 38302552 Free PMC article.
-
Identifying key targets and immune environment in wound healing based on iron overload-related genes.Arch Dermatol Res. 2025 Apr 19;317(1):719. doi: 10.1007/s00403-025-04140-y. Arch Dermatol Res. 2025. PMID: 40252113
References
-
- Clark J. M., Lambertsen C. J. Pulmonary oxygen toxicity: a review. Pharmacological Reviews . 1971;23(2):37–133. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
