

Novel trends of genome evolution in highly complex tropical sponge microbiomes
- PMID: 36195901
- PMCID: PMC9531527
- DOI: 10.1186/s40168-022-01359-z
Novel trends of genome evolution in highly complex tropical sponge microbiomes
Erratum in
-
Correction: Novel trends of genome evolution in highly complex tropical sponge microbiomes.Microbiome. 2022 Oct 24;10(1):182. doi: 10.1186/s40168-022-01393-x. Microbiome. 2022. PMID: 36280871 Free PMC article. No abstract available.
Abstract
Background: Tropical members of the sponge genus Ircinia possess highly complex microbiomes that perform a broad spectrum of chemical processes that influence host fitness. Despite the pervasive role of microbiomes in Ircinia biology, it is still unknown how they remain in stable association across tropical species. To address this question, we performed a comparative analysis of the microbiomes of 11 Ircinia species using whole-metagenomic shotgun sequencing data to investigate three aspects of bacterial symbiont genomes-the redundancy in metabolic pathways across taxa, the evolution of genes involved in pathogenesis, and the nature of selection acting on genes relevant to secondary metabolism.
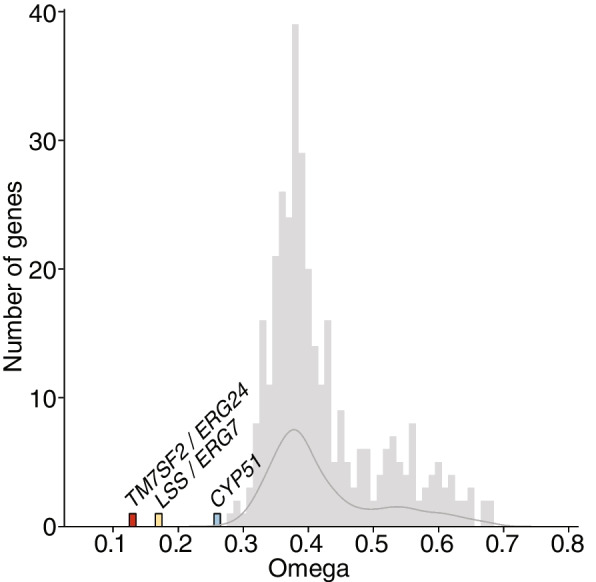
Results: A total of 424 new, high-quality bacterial metagenome-assembled genomes (MAGs) were produced for 10 Caribbean Ircinia species, which were evaluated alongside 113 publicly available MAGs sourced from the Pacific species Ircinia ramosa. Evidence of redundancy was discovered in that the core genes of several primary metabolic pathways could be found in the genomes of multiple bacterial taxa. Across hosts, the metagenomes were depleted in genes relevant to pathogenicity and enriched in eukaryotic-like proteins (ELPs) that likely mimic the hosts' molecular patterning. Finally, clusters of steroid biosynthesis genes (CSGs), which appear to be under purifying selection and undergo horizontal gene transfer, were found to be a defining feature of Ircinia metagenomes.
Conclusions: These results illustrate patterns of genome evolution within highly complex microbiomes that illuminate how associations with hosts are maintained. The metabolic redundancy within the microbiomes could help buffer the hosts from changes in the ambient chemical and physical regimes and from fluctuations in the population sizes of the individual microbial strains that make up the microbiome. Additionally, the enrichment of ELPs and depletion of LPS and cellular motility genes provide a model for how alternative strategies to virulence can evolve in microbiomes undergoing mixed-mode transmission that do not ultimately result in higher levels of damage (i.e., pathogenicity) to the host. Our last set of results provides evidence that sterol biosynthesis in Ircinia-associated bacteria is widespread and that these molecules are important for the survival of bacteria in highly complex Ircinia microbiomes. Video Abstract.
Keywords: Ircinia; Marine; Metagenomics; Microbiome; Sponge; Sterols; Tropical.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Vacelet J, Donadey C, Vacelet J, Donadey C. Electron microscope study of the association between some sponges and bacteria. J Exp Mar Biol Ecol. 1977;30:301–314. doi: 10.1016/0022-0981(77)90038-7. - DOI
-
- Wilkinson CR. Microbial associations in sponges. II. Numerical analysis of sponge and water bacterial populations. Mar Biol. 1978;49:169–176. doi: 10.1007/BF00387116. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
