

Virological characteristics of the SARS-CoV-2 Omicron BA.2 subvariants, including BA.4 and BA.5
- PMID: 36198317
- PMCID: PMC9472642
- DOI: 10.1016/j.cell.2022.09.018
Virological characteristics of the SARS-CoV-2 Omicron BA.2 subvariants, including BA.4 and BA.5
Abstract
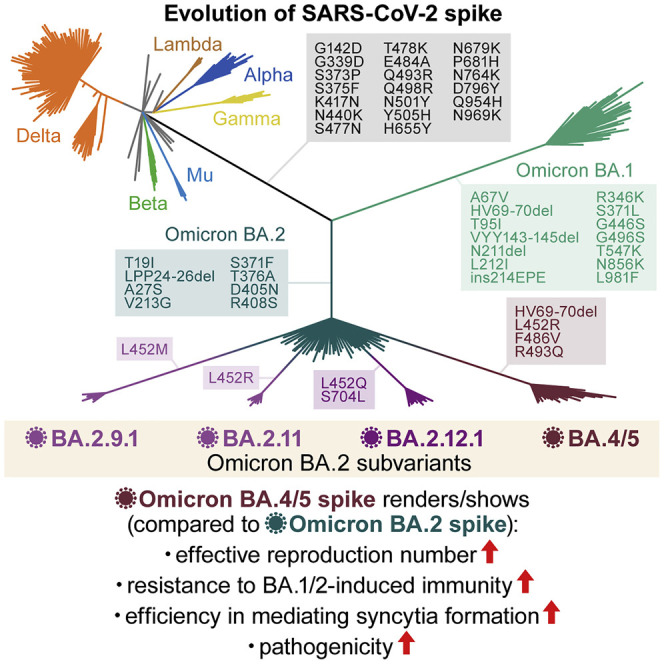
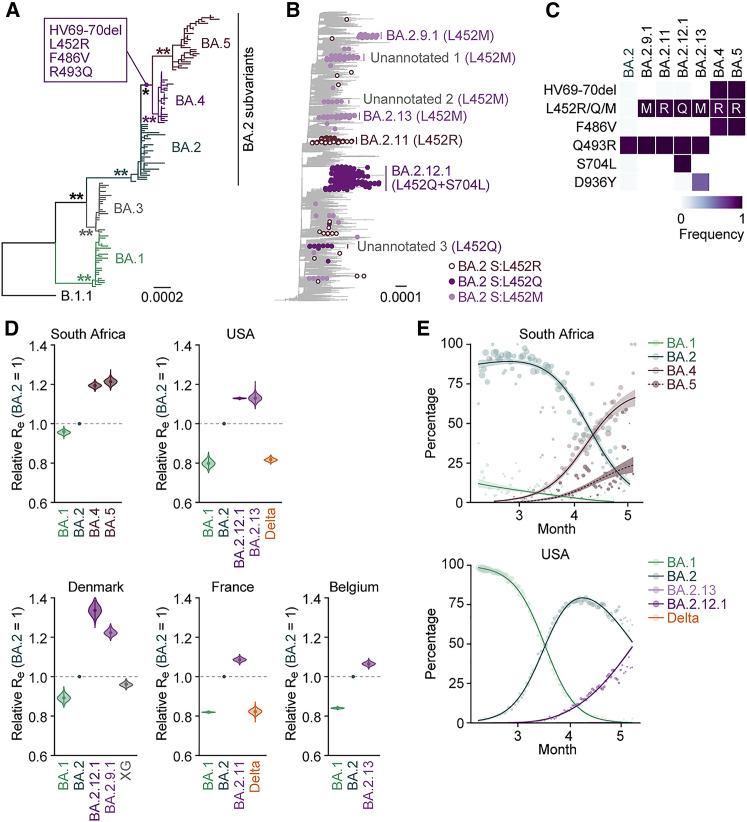
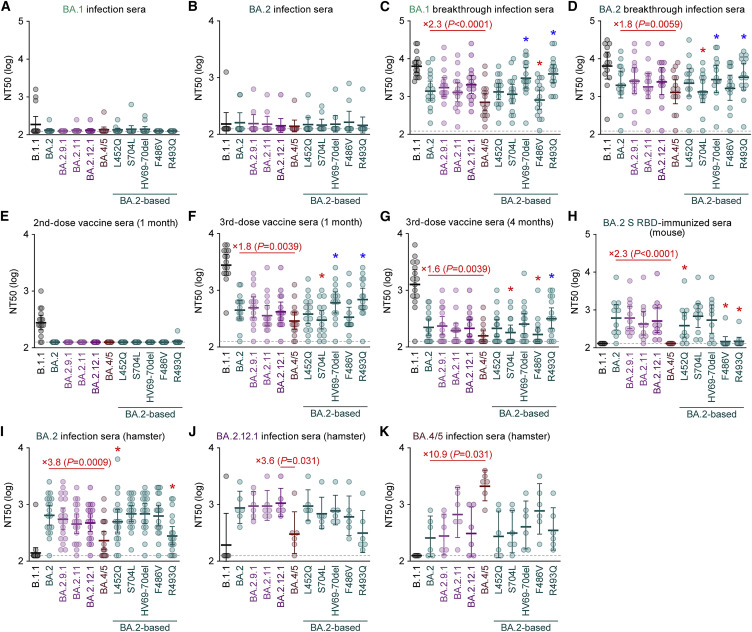
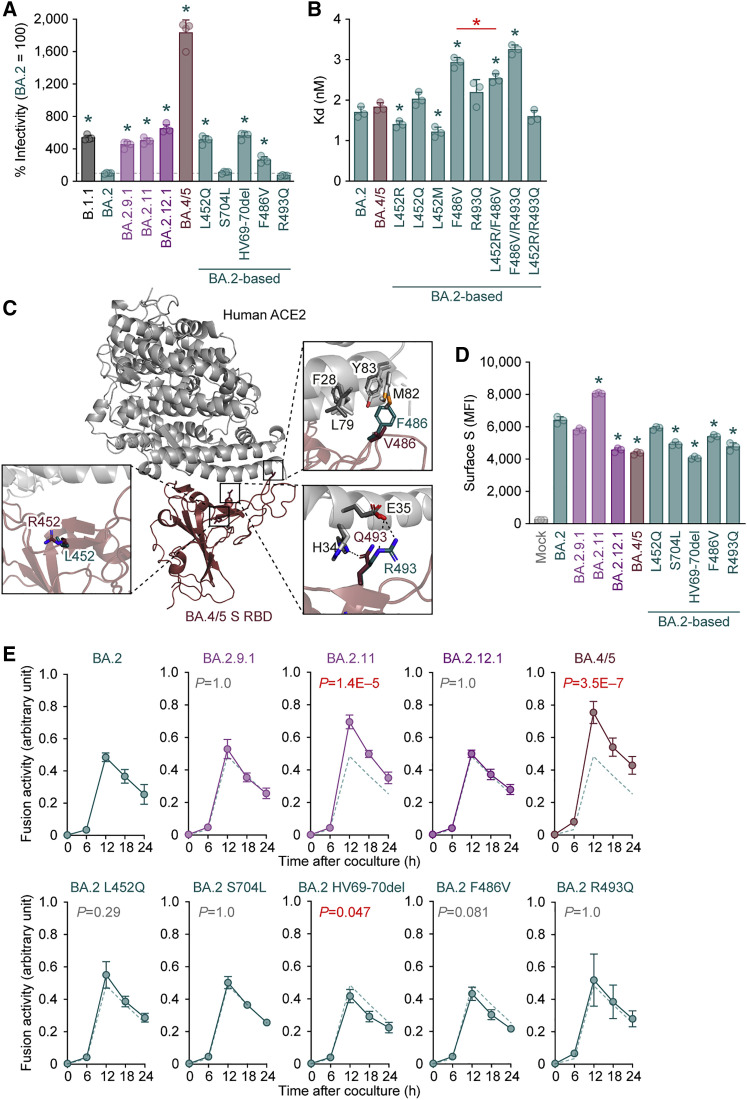
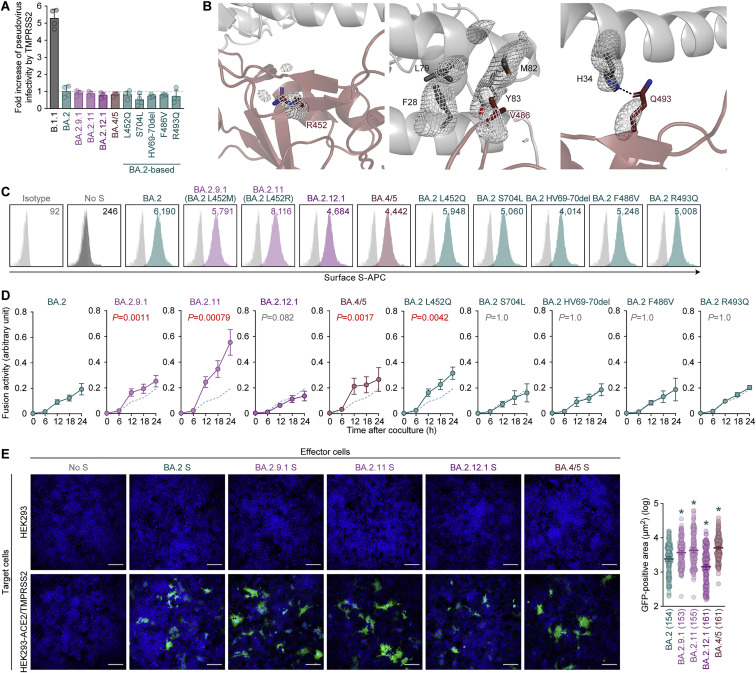
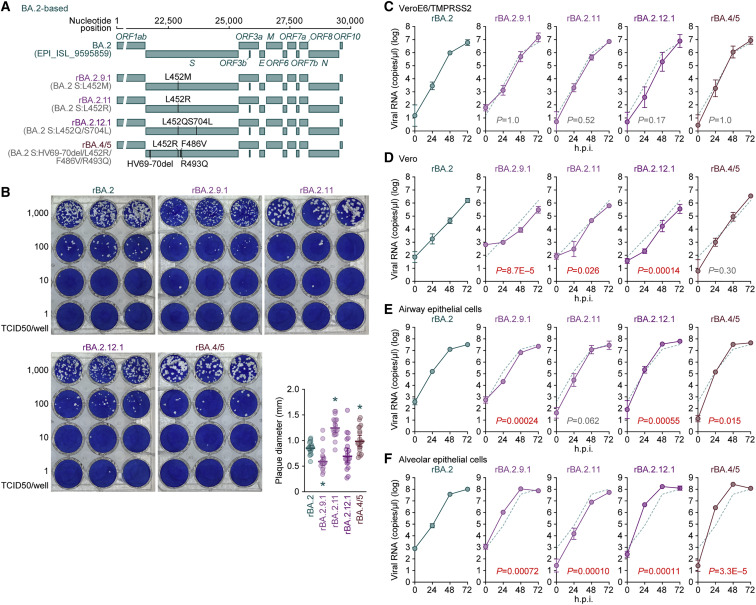
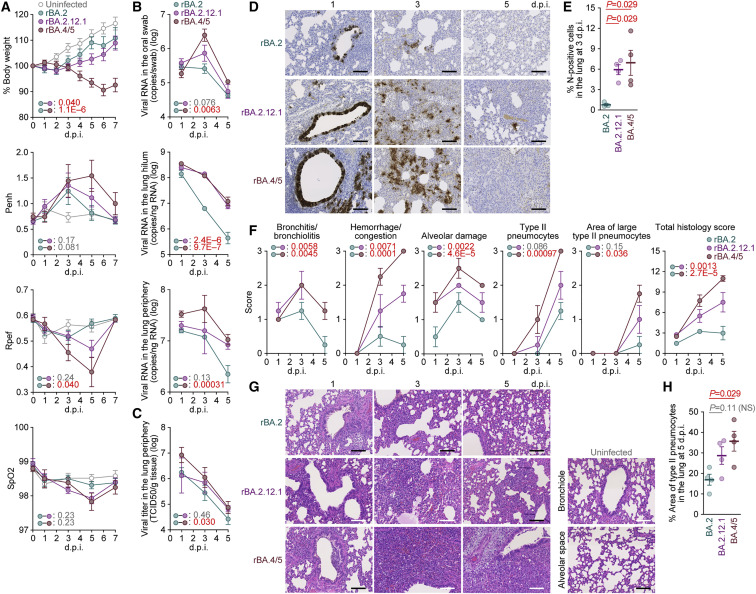
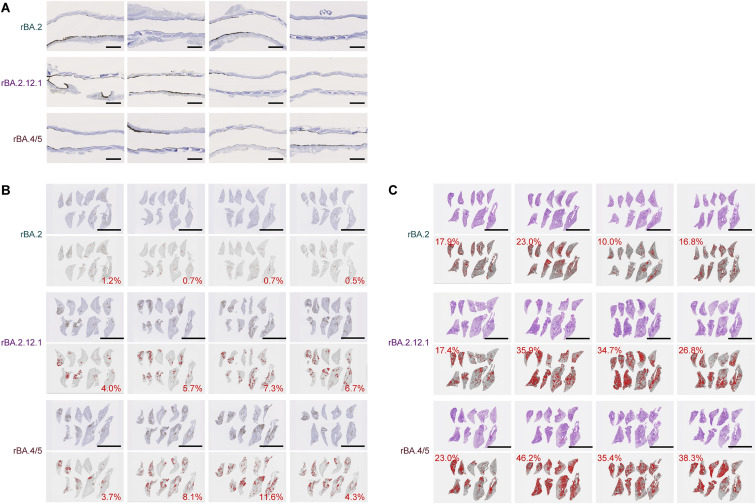
After the global spread of the SARS-CoV-2 Omicron BA.2, some BA.2 subvariants, including BA.2.9.1, BA.2.11, BA.2.12.1, BA.4, and BA.5, emerged in multiple countries. Our statistical analysis showed that the effective reproduction numbers of these BA.2 subvariants are greater than that of the original BA.2. Neutralization experiments revealed that the immunity induced by BA.1/2 infections is less effective against BA.4/5. Cell culture experiments showed that BA.2.12.1 and BA.4/5 replicate more efficiently in human alveolar epithelial cells than BA.2, and particularly, BA.4/5 is more fusogenic than BA.2. We further provided the structure of the BA.4/5 spike receptor-binding domain that binds to human ACE2 and considered how the substitutions in the BA.4/5 spike play roles in ACE2 binding and immune evasion. Moreover, experiments using hamsters suggested that BA.4/5 is more pathogenic than BA.2. Our multiscale investigations suggest that the risk of BA.2 subvariants, particularly BA.4/5, to global health is greater than that of original BA.2.
Keywords: BA.2; BA.2.12.1; BA.4; BA.5; COVID-19; Omicron; SARS-CoV-2; immune resistance; pathogenicity; transmissibility.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Y.Y. and T.N. are founders and shareholders of HiLung, Inc. J.K. is an employee of HiLung, Inc. Y.Y. is a co-inventor of patents (PCT/JP2016/057254; “Method for inducing differentiation of alveolar epithelial cells,” PCT/JP2016/059786, “Method of producing airway epithelial cells”).
Figures

References
-
- Abbas Q., Kusakin A., Sharrouf K., Jyakhwo S., Komissarov A.S. 2022. Follow-up investigation and detailed mutational characterization of the SARS-CoV-2 Omicron variant lineages (BA.1, BA.2, BA.3 and BA.1.1) - DOI
-
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. - DOI - PMC - PubMed
-
- Aggarwal A., Akerman A., Milogiannakis V., Silva M.R., Walker G., Kidinger A., Angelovich T., Waring E., Amatayakul-Chantler S., Roth N., et al. SARS-CoV-2 omicron BA.5: evolving tropism and evasion of potent humoral responses and resistance to clinical immunotherapeutics relative to viral variants of concern. eBioMedicine. 2022;84:104270. doi: 10.1016/j.ebiom.2022.104270. - DOI - PMC - PubMed
-
- Altarawneh H.N., Chemaitelly H., Ayoub H.H., Hasan M.R., Coyle P., Yassine H.M., Al-Khatib H.A., Benslimane F.M., Al-Kanaani Z., Al-Kuwari E., et al. 2022. Protection of SARS-CoV-2 natural infection against reinfection with the Omicron BA.4 or BA.5 subvariants. - DOI
-
- Aricescu A.R., Assenberg R., Bill R.M., Busso D., Chang V.T., Davis S.J., Dubrovsky A., Gustafsson L., Hedfalk K., Heinemann U., et al. Eukaryotic expression: developments for structural proteomics. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1114–1124. doi: 10.1107/S0907444906029805. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
