

Genomic Epidemiology and Phylodynamic Analysis of Enterovirus A71 Reveal Its Transmission Dynamics in Asia
- PMID: 36200890
- PMCID: PMC9603238
- DOI: 10.1128/spectrum.01958-22
Genomic Epidemiology and Phylodynamic Analysis of Enterovirus A71 Reveal Its Transmission Dynamics in Asia
Abstract
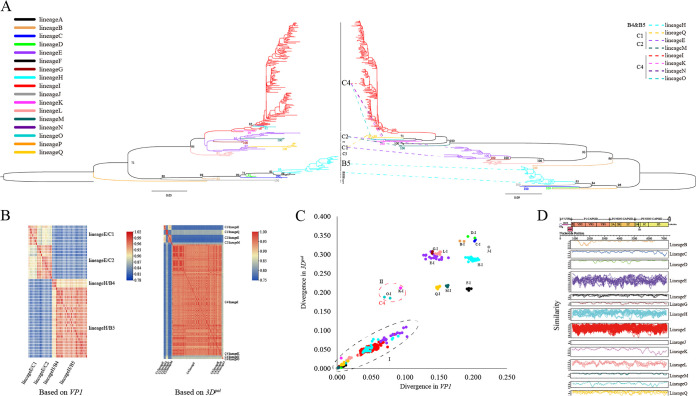
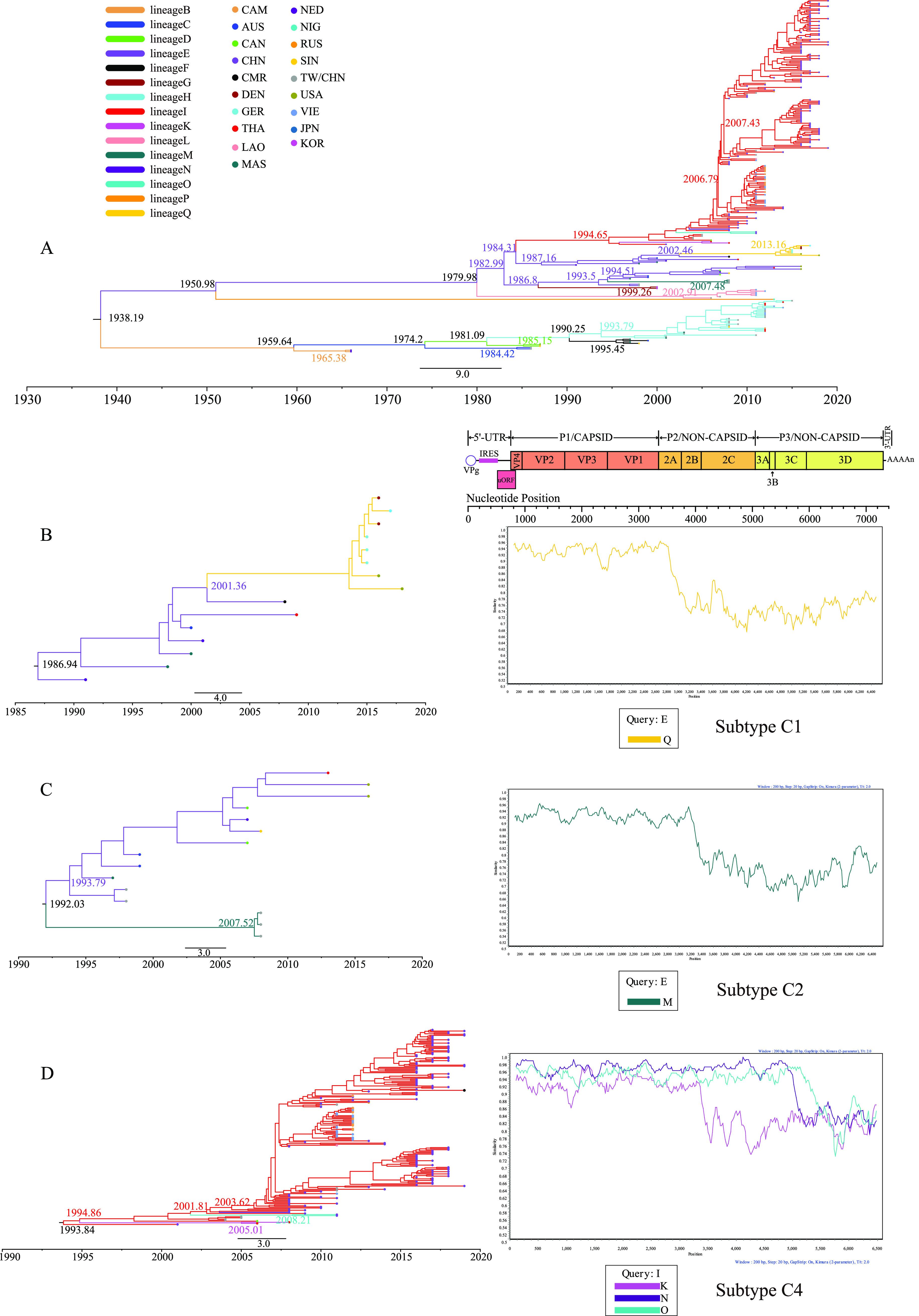
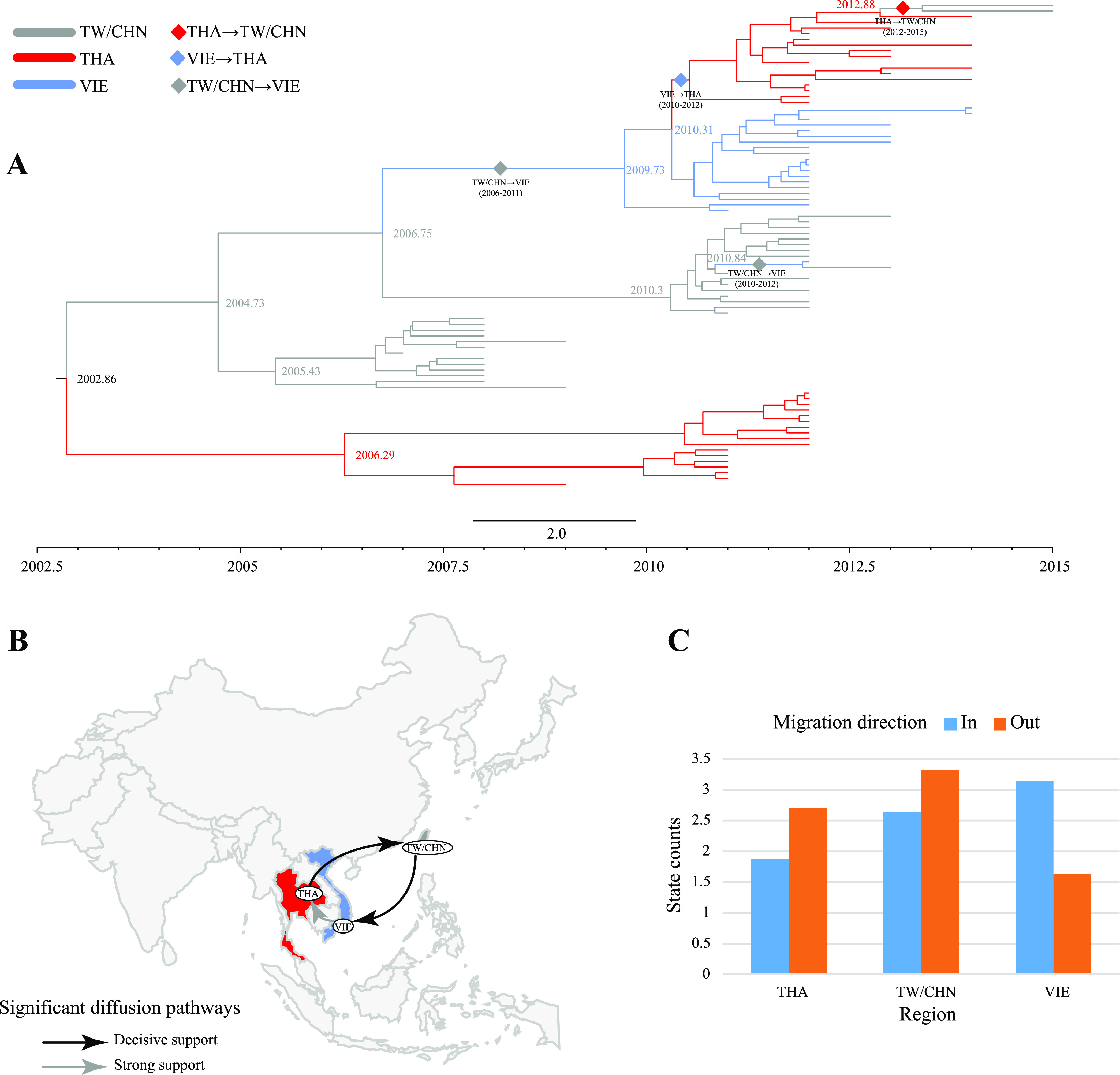
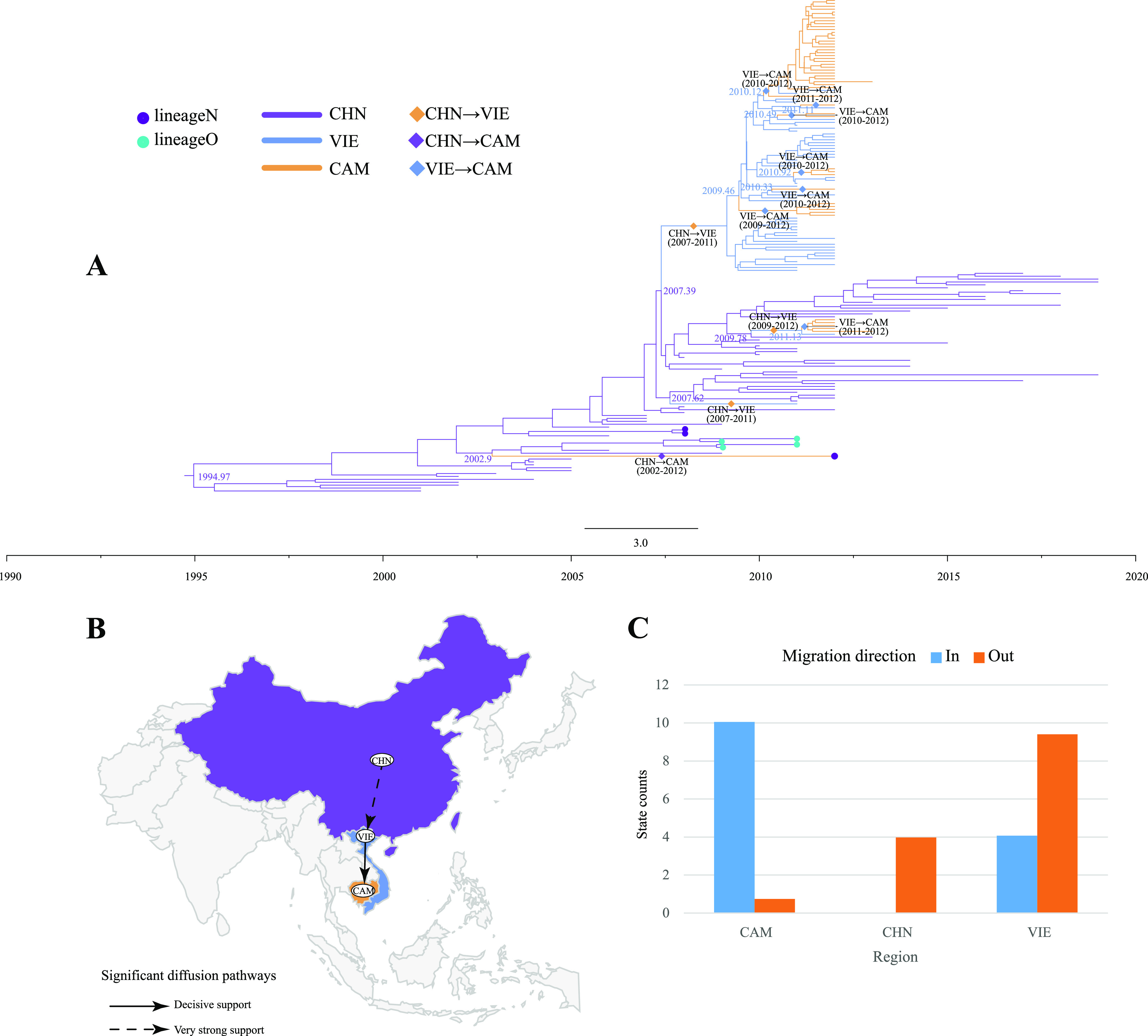
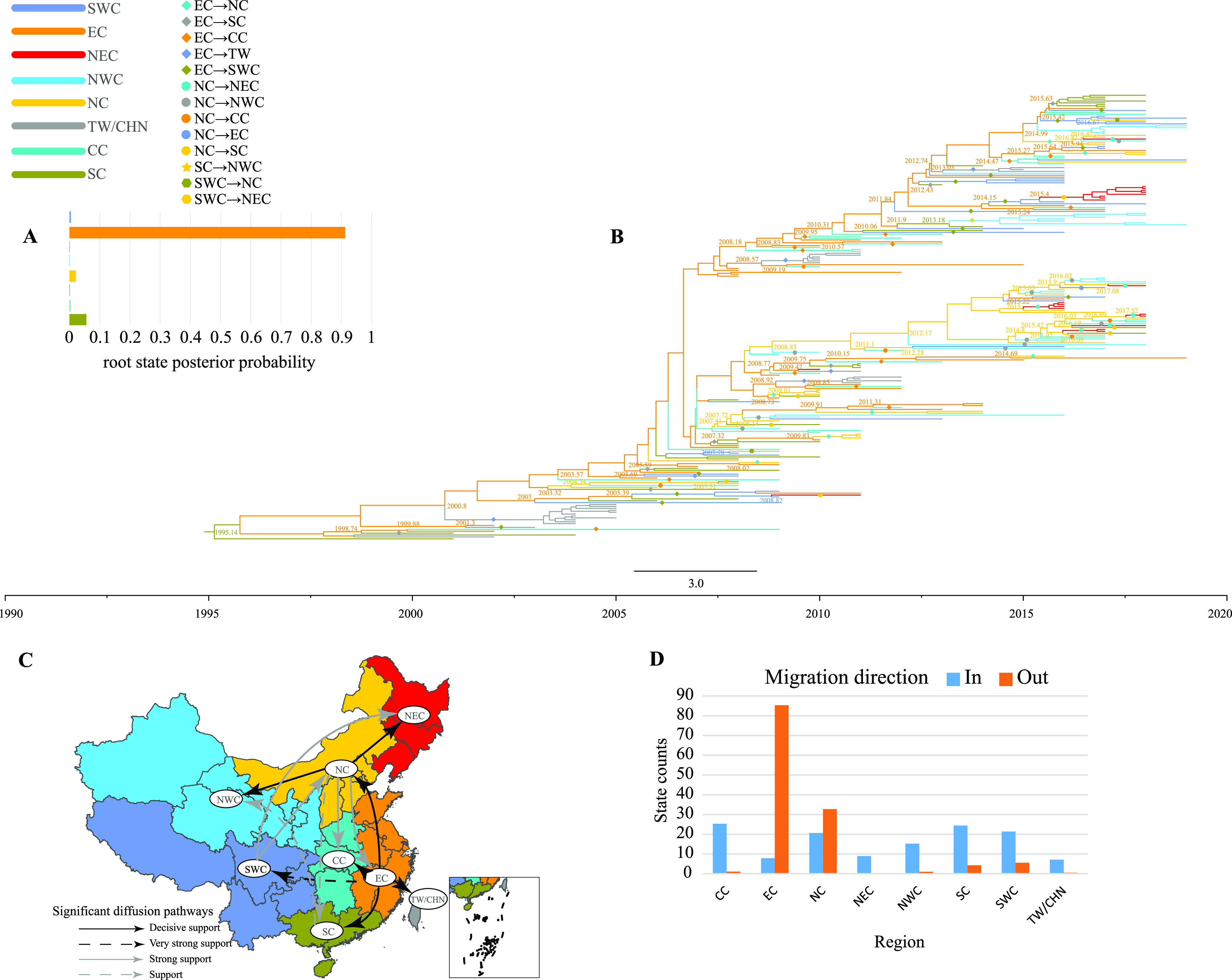
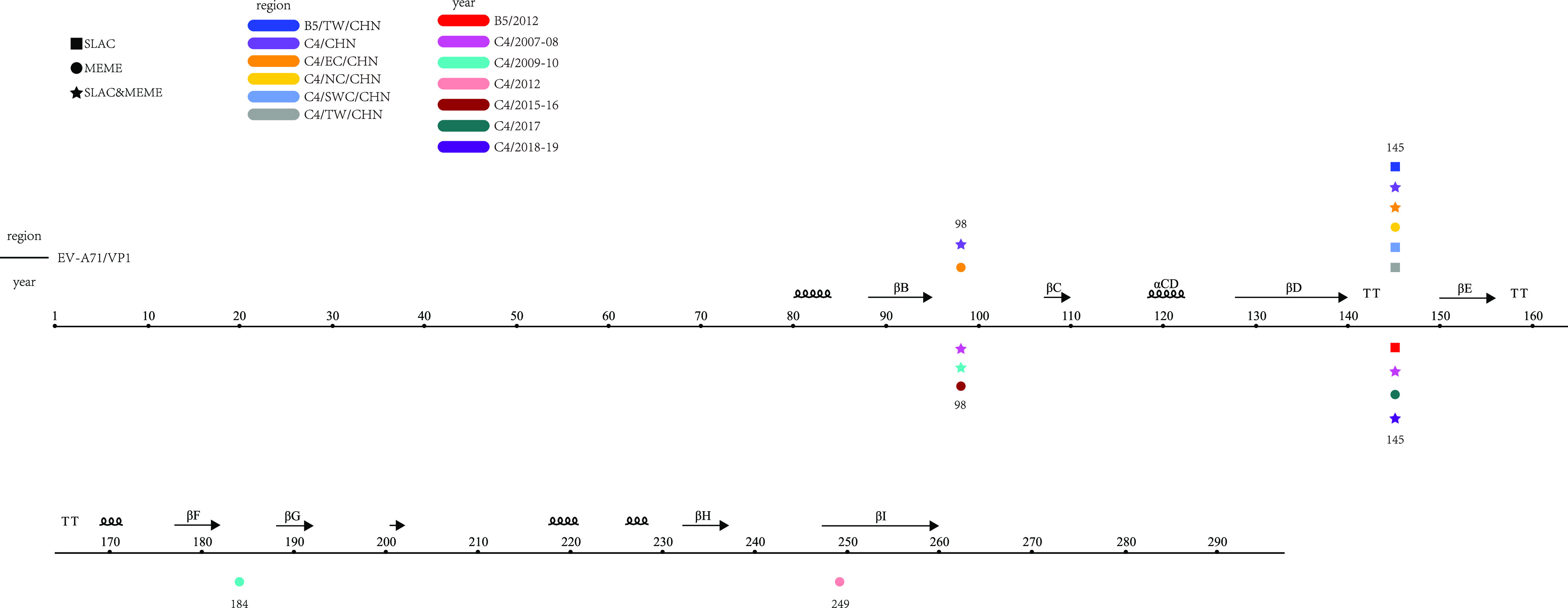
Enterovirus A71 (EV-A71) is one of the main pathogens causing hand, foot, and mouth disease (HFMD) outbreaks in Asian children under 5 years of age. In severe cases, it can cause neurological complications and be life-threatening. In this study, 200 newly sequenced EV-A71 whole-genome sequences were combined with 772 EV-A71 sequences from GenBank for large-scale analysis to investigate global EV-A71 epidemiology, phylogeny, and Bayesian phylodynamic characteristics. Based on the phylogenetic analysis of the EV-A71 3Dpol region, six new evolutionary lineages (lineages B, J, K, O, P, and Q) were found in this study, and the number of evolutionary lineages was expanded from 11 to 17. Temporal dynamics and recombination breakpoint analyses based on genotype C revealed that recombination of nonstructural protein-coding regions, including 3Dpol, is an important reason for the emergence of new lineages. The EV-A71 epidemic in the Asia-Pacific region is complex, and phylogeographic analysis found that Vietnam played a key role in the spread of subgenotypes B5 and C4. The origin of EV-A71 subgenotype C4 in China is East China, which is closely related to the prevalence of subgenotype C4 in the south and throughout China. Selection pressure analysis revealed that, in addition to VP1 amino acid residues VP1-98 and VP1-145, which are associated with EV-A71 pathogenicity, amino acid residues VP1-184 and VP1-249 were also positively selected, and their functions still need to be determined by biology and immunology. This study aimed to provide a solid theoretical basis for EV-A71-related disease surveillance and prevention, antiviral research, and vaccine development through a comprehensive analysis. IMPORTANCE EV-A71 is one of the most important pathogens causing HFMD outbreaks; however, large-scale studies of EV-A71 genomic epidemiology are currently lacking. In this study, 200 new EV-A71 whole-genome sequences were determined. Combining these with 772 EV-A71 whole-genome sequences in the GenBank database, the evolutionary and transmission characteristics of global and Asian EV-A71 were analyzed. Six new evolutionary lineages were identified in this study. We also found that recombination in nonstructural protein-coding regions, including 3Dpol, is an important cause for the emergence of new lineages. The results provided a solid theoretical basis for EV-A71-related disease surveillance and prevention, antiviral research, and vaccine development.
Keywords: Bayesian phylodynamics; EV-A71; hand foot and mouth disease; phylogenetics; recombination.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lulla V, Dinan AM, Hosmillo M, Chaudhry Y, Sherry L, Irigoyen N, Nayak KM, Stonehouse NJ, Zilbauer M, Goodfellow I, Firth AE. 2019. An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells. Nat Microbiol 4:280–292. doi: 10.1038/s41564-018-0297-1. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
