

Dynamic allostery in substrate binding by human thymidylate synthase
- PMID: 36200982
- PMCID: PMC9536839
- DOI: 10.7554/eLife.79915
Dynamic allostery in substrate binding by human thymidylate synthase
Abstract
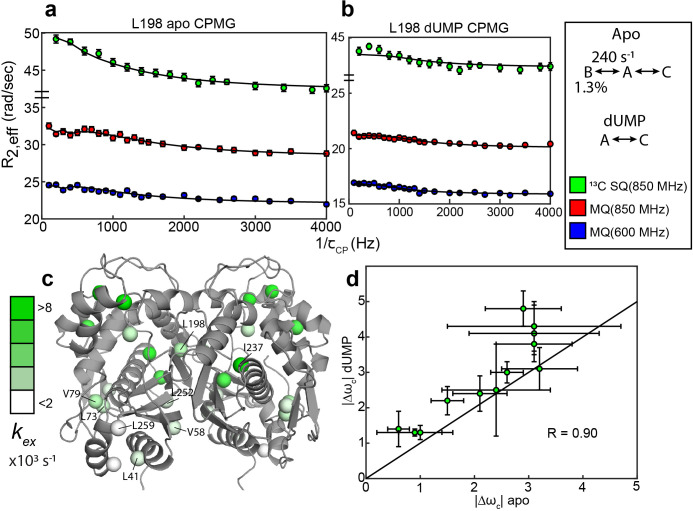
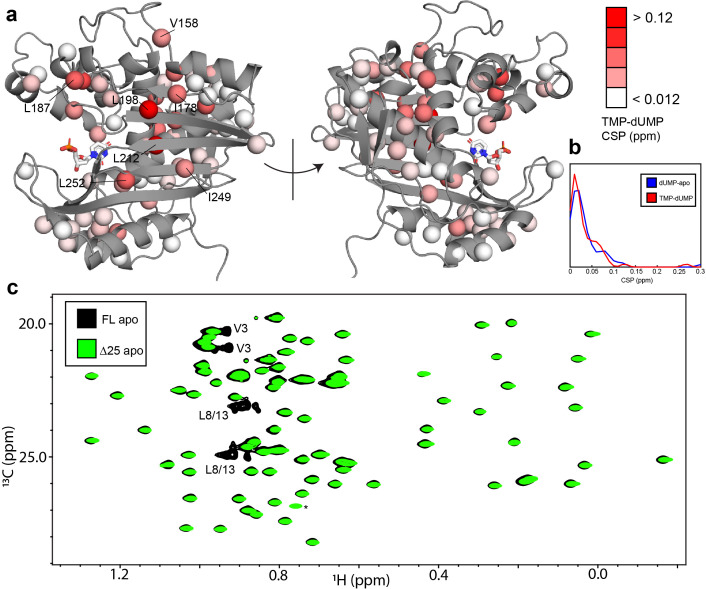
Human thymidylate synthase (hTS) is essential for DNA replication and therefore a therapeutic target for cancer. Effective targeting requires knowledge of the mechanism(s) of regulation of this 72 kDa homodimeric enzyme. Here, we investigate the mechanism of binding cooperativity of the nucleotide substrate. We have employed exquisitely sensitive methyl-based CPMG and CEST NMR experiments enabling us to identify residues undergoing bifurcated linear 3-state exchange, including concerted switching between active and inactive conformations in the apo enzyme. The inactive state is populated to only ~1.3%, indicating that conformational selection contributes negligibly to the cooperativity. Instead, methyl rotation axis order parameters, determined by 2H transverse relaxation rates, suggest that rigidification of the enzyme upon substrate binding is responsible for the entropically-driven cooperativity. Lack of the rigidification in product binding and substrate binding to an N-terminally truncated enzyme, both non-cooperative, support this idea. In addition, the lack of this rigidification in the N-terminal truncation indicates that interactions between the flexible N-terminus and the rest of the protein, which are perturbed by substrate binding, play a significant role in the cooperativity-a novel mechanism of dynamic allostery. Together, these findings yield a rare depth of insight into the substrate binding cooperativity of an essential enzyme.
Keywords: NMR; allostery; conformational entropy; cooperativity; human; molecular biophysics; protein dynamics; structural biology; thymidylate synthase.
© 2022, Bonin et al.
Conflict of interest statement
JB, PS, AL No competing interests declared
Figures
















References
-
- Baker WN. A new comparison of the viscosity of D2O with that of H2O. Journal of Chemical Physics. 1936;4:294. doi: 10.1063/1.1749842. - DOI
-
- Bernini A, Venditti V, Spiga O, Niccolai N. Probing protein surface accessibility with solvent and paramagnetic molecules. Progress in Nuclear Magnetic Resonance Spectroscopy. 2009;54:278–289. doi: 10.1016/j.pnmrs.2008.10.003. - DOI
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
