

Contribution of adaptive immunity to human COPD and experimental models of emphysema
- PMID: 36201635
- PMCID: PMC9886356
- DOI: 10.1152/physrev.00036.2021
Contribution of adaptive immunity to human COPD and experimental models of emphysema
Abstract
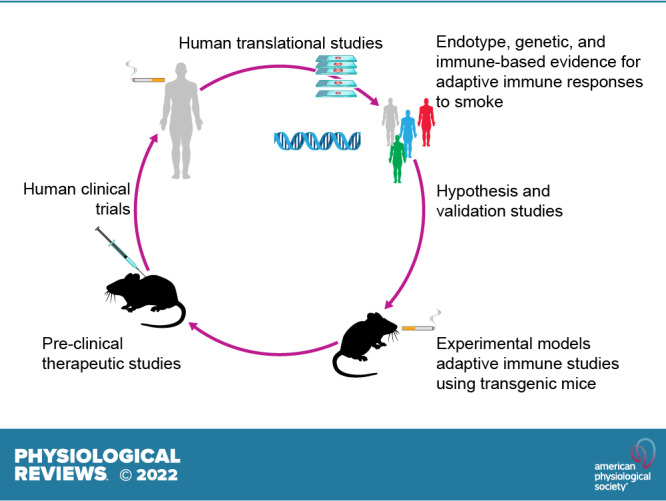
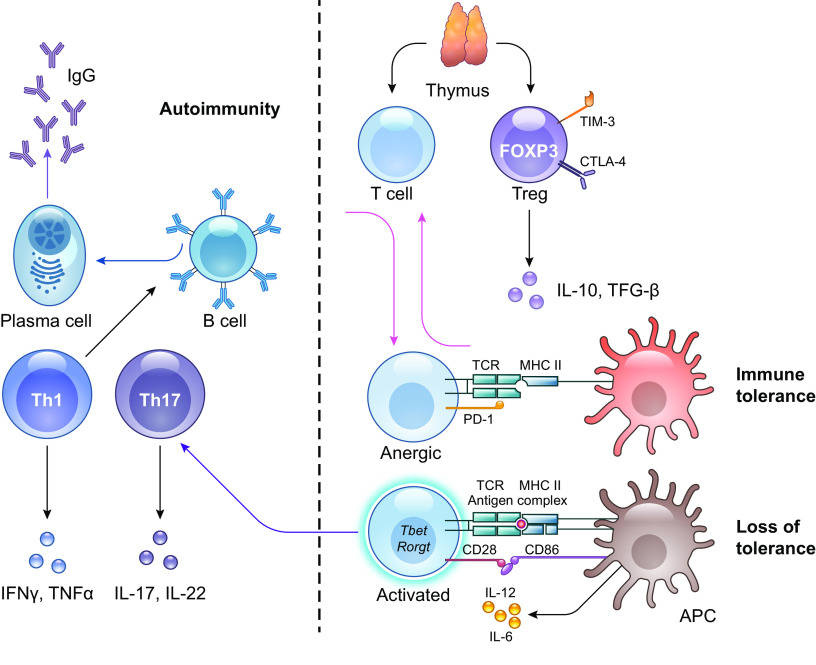
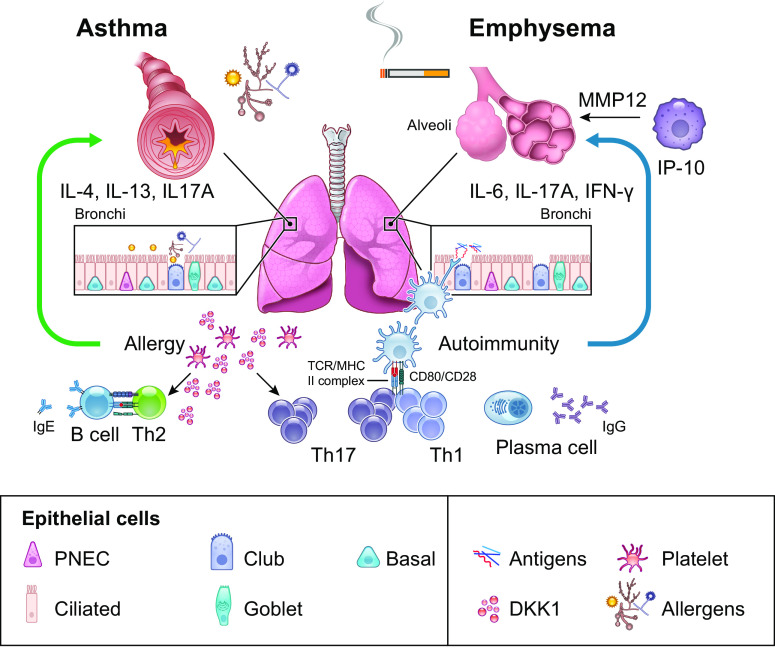
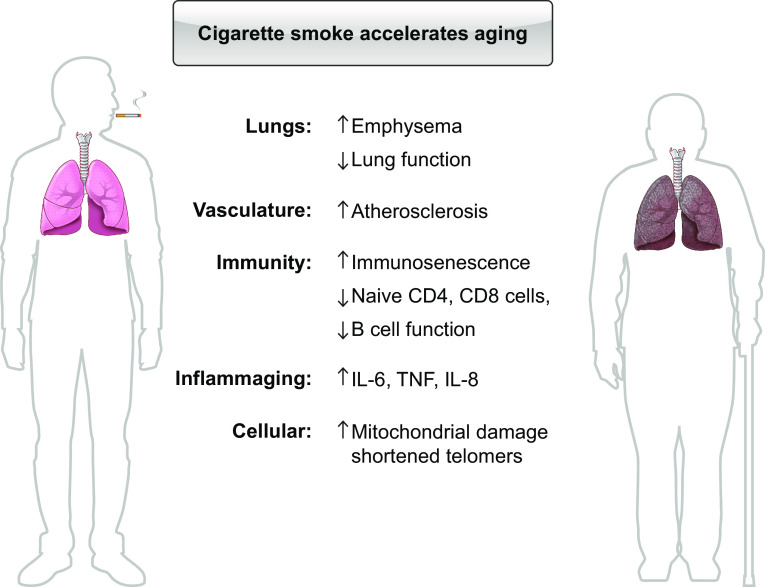
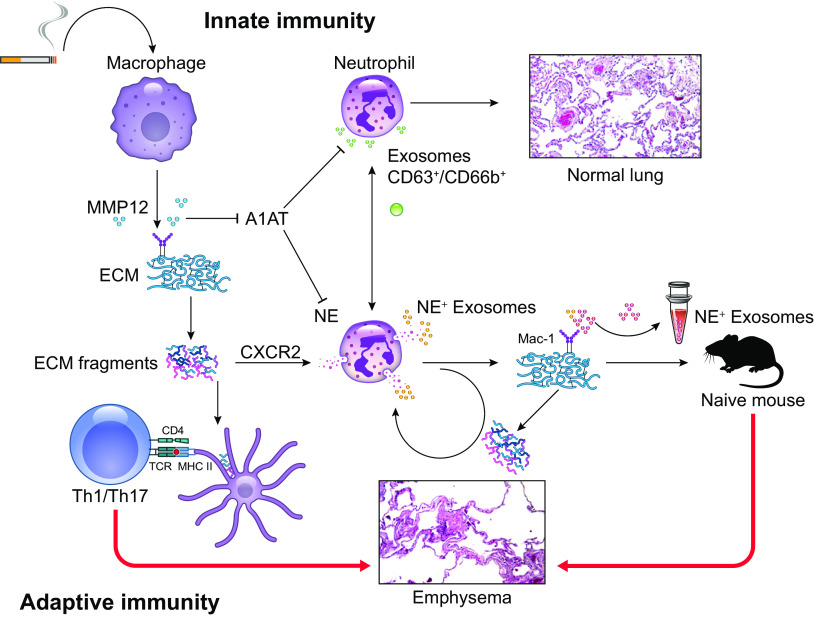
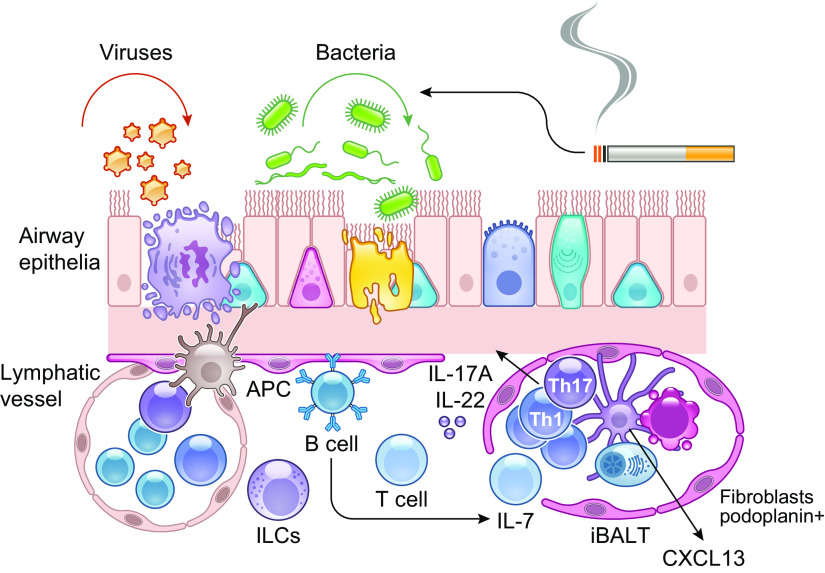
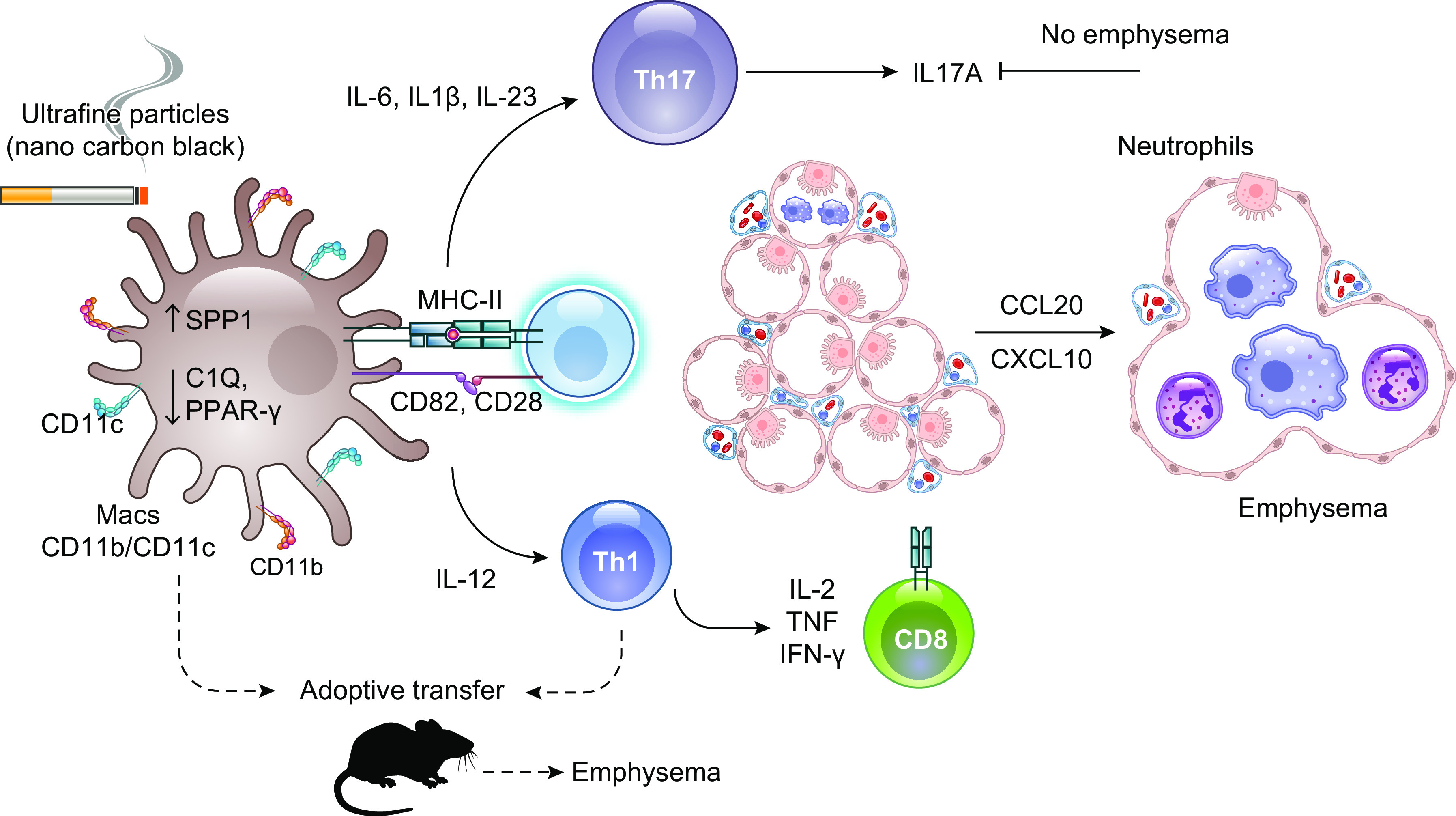
The pathophysiology of chronic obstructive pulmonary disease (COPD) and the undisputed role of innate immune cells in this condition have dominated the field in the basic research arena for many years. Recently, however, compelling data suggesting that adaptive immune cells may also contribute to the progressive nature of lung destruction associated with COPD in smokers have gained considerable attention. The histopathological changes in the lungs of smokers can be limited to the large or small airways, but alveolar loss leading to emphysema, which occurs in some individuals, remains its most significant and irreversible outcome. Critically, however, the question of why emphysema progresses in a subset of former smokers remained a mystery for many years. The recognition of activated and organized tertiary T- and B-lymphoid aggregates in emphysematous lungs provided the first clue that adaptive immune cells may play a crucial role in COPD pathophysiology. Based on these findings from human translational studies, experimental animal models of emphysema were used to determine the mechanisms through which smoke exposure initiates and orchestrates adaptive autoreactive inflammation in the lungs. These models have revealed that T helper (Th)1 and Th17 subsets promote a positive feedback loop that activates innate immune cells, confirming their role in emphysema pathogenesis. Results from genetic studies and immune-based discoveries have further provided strong evidence for autoimmunity induction in smokers with emphysema. These new findings offer a novel opportunity to explore the mechanisms underlying the inflammatory landscape in the COPD lung and offer insights for development of precision-based treatment to halt lung destruction.
Keywords: T lymphocytes; adaptive immunity; autoimmunity; emphysema; exacerbation.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- Wilson RW. Cigarette smoking, disability days and respiratory conditions. J Occup Med 15: 236–240, 1973. - PubMed
-
- Institute for Health Metrics and Evaluation. Findings from the Global Burden of Disease Study 2017. Seattle, WA: IHME, 2018.
-
- de Koning HJ, van der Aalst CM, de Jong PA, Scholten ET, Nackaerts K, Heuvelmans MA, Lammers JJ, Weenink C, Yousaf-Khan U, Horeweg N, van 't Westeinde S, Prokop M, Mali WP, Mohamed Hoesein FA, van Ooijen PM, Aerts J, den Bakker MA, Thunnissen E, Verschakelen J, Vliegenthart R, Walter JE, Ten Haaf K, Groen HJ, Oudkerk M. Reduced lung-cancer mortality with volume CT screening in a randomized trial. N Engl J Med 382: 503–513, 2020. doi: 10.1056/NEJMoa1911793. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical