

Quasiparticle Self-Consistent GW-Bethe-Salpeter Equation Calculations for Large Chromophoric Systems
- PMID: 36201788
- PMCID: PMC9648197
- DOI: 10.1021/acs.jctc.2c00531
Quasiparticle Self-Consistent GW-Bethe-Salpeter Equation Calculations for Large Chromophoric Systems
Abstract
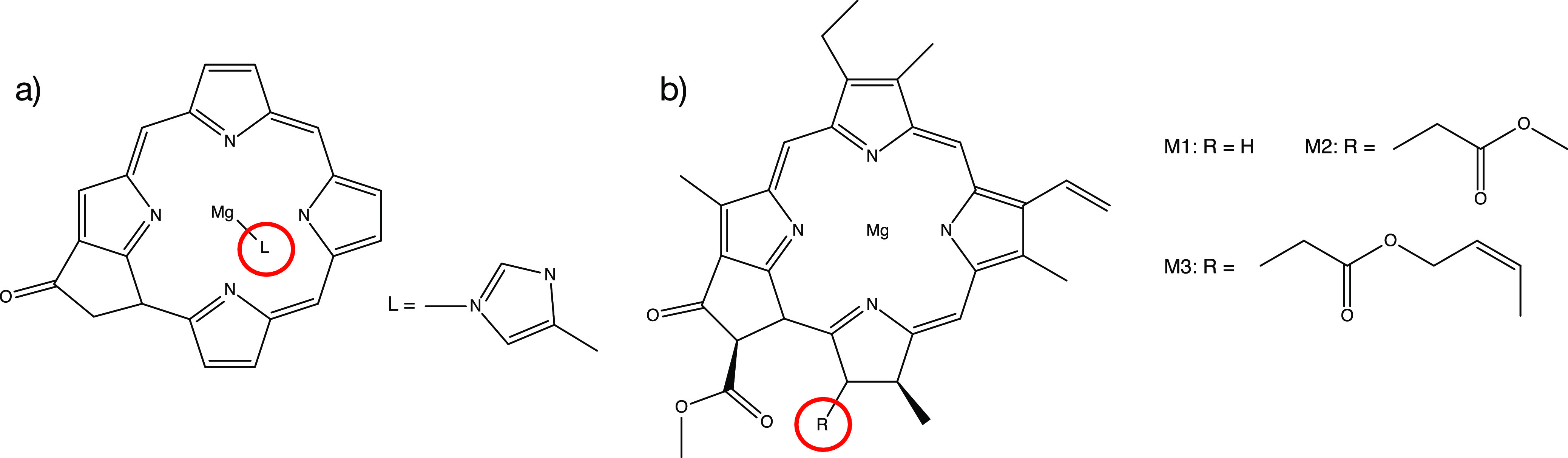
The GW-Bethe-Salpeter equation (BSE) method is promising for calculating the low-lying excitonic states of molecular systems. However, so far it has only been applied to rather small molecules and in the commonly implemented diagonal approximations to the electronic self-energy, it depends on a mean-field starting point. We describe here an implementation of the self-consistent and starting-point-independent quasiparticle self-consistent (qsGW)-BSE approach, which is suitable for calculations on large molecules. We herein show that eigenvalue-only self-consistency can lead to an unfaithful description of some excitonic states for chlorophyll dimers while the qsGW-BSE vertical excitation energies (VEEs) are in excellent agreement with spectroscopic experiments for chlorophyll monomers and dimers measured in the gas phase. Furthermore, VEEs from time-dependent density functional theory calculations tend to disagree with experimental values and using different range-separated hybrid (RSH) kernels does change the VEEs by up to 0.5 eV. We use the new qsGW-BSE implementation to calculate the lowest excitation energies of the six chromophores of the photosystem II (PSII) reaction center (RC) with nearly 2000 correlated electrons. Using more than 11,000 (6000) basis functions, the calculation could be completed in less than 5 (2) days on a single modern compute node. In agreement with previous TD-DFT calculations using RSH kernels on models that also do not include environmental effects, our qsGW-BSE calculations only yield states with local characters in the low-energy spectrum of the hexameric complex. Earlier works with RSH kernels have demonstrated that the protein environment facilitates the experimentally observed interchromophoric charge transfer. Therefore, future research will need to combine correlation effects beyond TD-DFT with an explicit treatment of environmental electrostatics.
Conflict of interest statement
The authors declare no competing financial interest.
Figures


Similar articles
-
Accuracy Assessment of GW Starting Points for Calculating Molecular Excitation Energies Using the Bethe-Salpeter Formalism.J Chem Theory Comput. 2018 Apr 10;14(4):2127-2136. doi: 10.1021/acs.jctc.8b00014. Epub 2018 Mar 15. J Chem Theory Comput. 2018. PMID: 29499116
-
Assessing the Role of the Kohn-Sham Density in the Calculation of the Low-Lying Bethe-Salpeter Excitation Energies.J Phys Chem A. 2023 Mar 23;127(11):2618-2627. doi: 10.1021/acs.jpca.2c07526. Epub 2023 Mar 13. J Phys Chem A. 2023. PMID: 36913525
-
An optimally tuned range-separated hybrid starting point for ab initio GW plus Bethe-Salpeter equation calculations of molecules.J Chem Phys. 2022 Aug 21;157(7):074103. doi: 10.1063/5.0097582. J Chem Phys. 2022. PMID: 35987597
-
A Guide to Molecular Properties from the Bethe-Salpeter Equation.J Phys Chem Lett. 2025 Apr 24;16(16):3980-3990. doi: 10.1021/acs.jpclett.5c00494. Epub 2025 Apr 14. J Phys Chem Lett. 2025. PMID: 40227071 Review.
-
Excited states properties of organic molecules: from density functional theory to the GW and Bethe-Salpeter Green's function formalisms.Philos Trans A Math Phys Eng Sci. 2014 Feb 10;372(2011):20130271. doi: 10.1098/rsta.2013.0271. Print 2014 Mar 13. Philos Trans A Math Phys Eng Sci. 2014. PMID: 24516185 Review.
Cited by
-
Toward Pair Atomic Density Fitting for Correlation Energies with Benchmark Accuracy.J Chem Theory Comput. 2023 Mar 14;19(5):1499-1516. doi: 10.1021/acs.jctc.2c01201. Epub 2023 Feb 14. J Chem Theory Comput. 2023. PMID: 36787494 Free PMC article.
-
Low-Scaling GW Algorithm Applied to Twisted Transition-Metal Dichalcogenide Heterobilayers.J Chem Theory Comput. 2024 Mar 12;20(5):2202-2208. doi: 10.1021/acs.jctc.3c01230. Epub 2024 Feb 14. J Chem Theory Comput. 2024. PMID: 38353944 Free PMC article.
-
Electronic Couplings and Conversion Dynamics between Localized and Charge Transfer Excitations from Many-Body Green's Functions Theory.J Chem Theory Comput. 2024 Jun 11;20(11):4605-4615. doi: 10.1021/acs.jctc.4c00142. Epub 2024 May 21. J Chem Theory Comput. 2024. PMID: 38770562 Free PMC article.
-
Reference Energies for Valence Ionizations and Satellite Transitions.J Chem Theory Comput. 2024 Jun 11;20(11):4751-4777. doi: 10.1021/acs.jctc.4c00216. Epub 2024 May 22. J Chem Theory Comput. 2024. PMID: 38776293 Free PMC article.
-
Beyond Quasi-Particle Self-Consistent GW for Molecules with Vertex Corrections.J Chem Theory Comput. 2025 Feb 25;21(4):1709-1721. doi: 10.1021/acs.jctc.4c01639. Epub 2025 Feb 11. J Chem Theory Comput. 2025. PMID: 39930976 Free PMC article.
References
-
- Kasha M.; Rawls H. R.; Ashraf El-Bayoumi M. A. The Exciton Model In Molecular Spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. 10.1351/pac196511030371. - DOI
-
- Reimers J. R.; Biczysko M.; Bruce D.; Coker D. F.; Frankcombe T. J.; Hashimoto H.; Hauer J.; Jankowiak R.; Kramer T.; Linnanto J.; Mamedov F.; Müh F.; Rätsep M.; Renger T.; Styring S.; Wan J.; Wang Z.; Wang-Otomo Z. Y.; Weng Y. X.; Yang C.; Zhang J. P.; Freiberg A.; Krausz E. Challenges facing an understanding of the nature of low-energy excited states in photosynthesis. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 1627–1640. 10.1016/j.bbabio.2016.06.010. - DOI - PubMed
-
- Yin S.; Dahlbom M. G.; Canfield P. J.; Hush N. S.; Kobayashi R.; Reimers J. R. Assignment of the Qy, absorption spectrum of photosystem-i from thermosynechococcus elongatus based on CAM-B3LYP calculations at the PW91-optimized protein structure. J. Phys. Chem. B 2007, 111, 9923–9930. 10.1021/jp070030p. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
