

Monkeypox virus: The changing facets of a zoonotic pathogen
- PMID: 36202208
- PMCID: PMC9534092
- DOI: 10.1016/j.meegid.2022.105372
Monkeypox virus: The changing facets of a zoonotic pathogen
Abstract
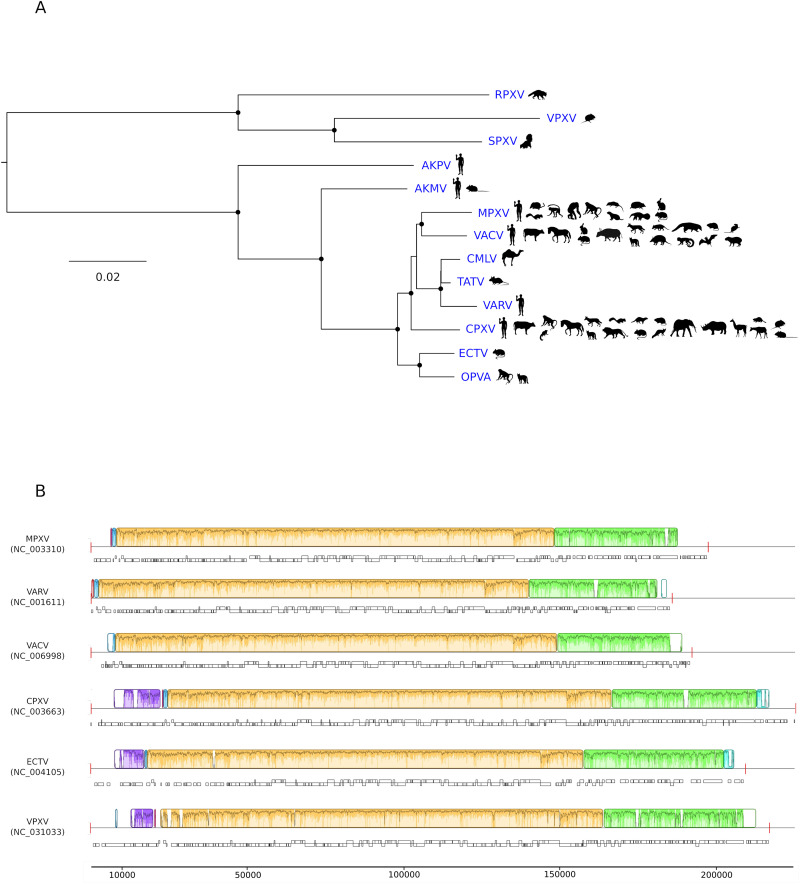
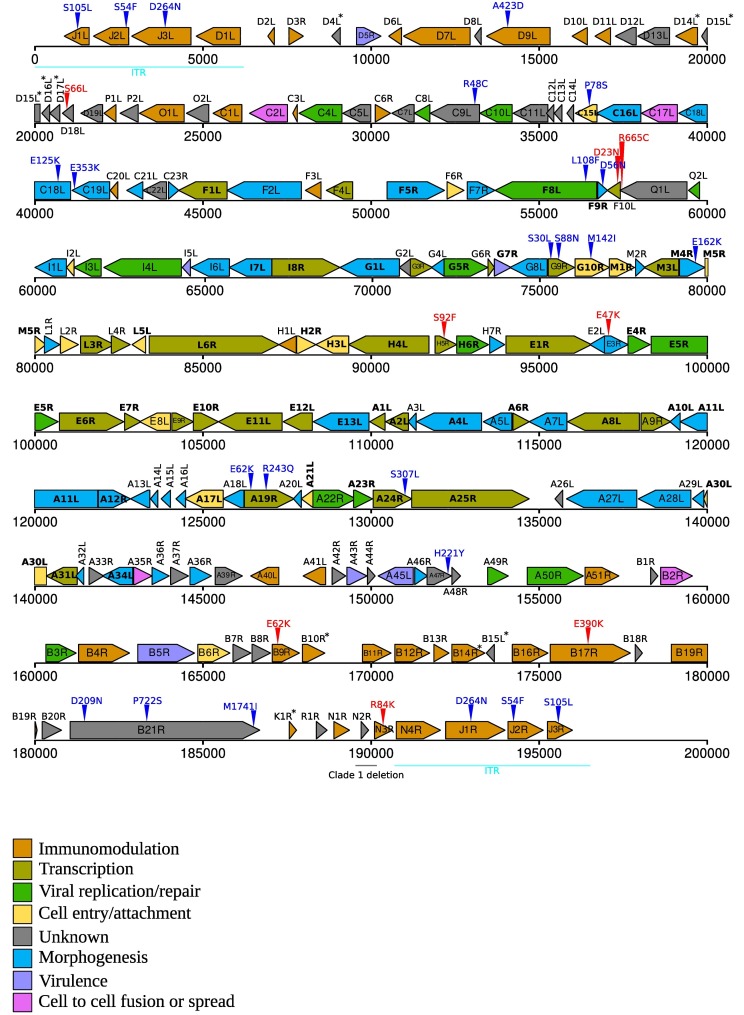
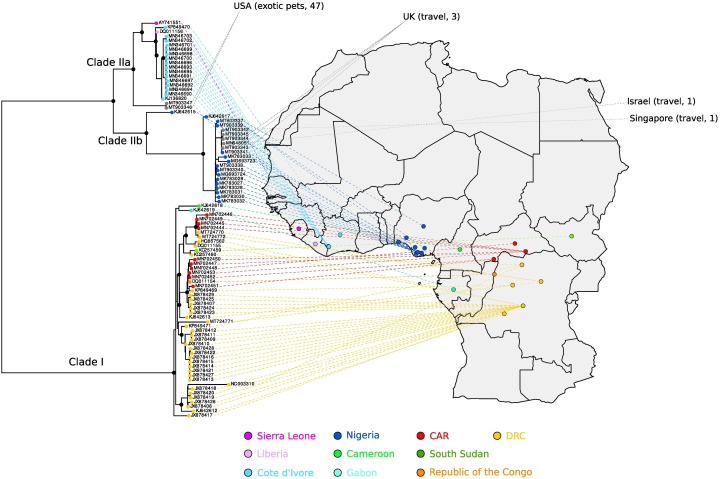
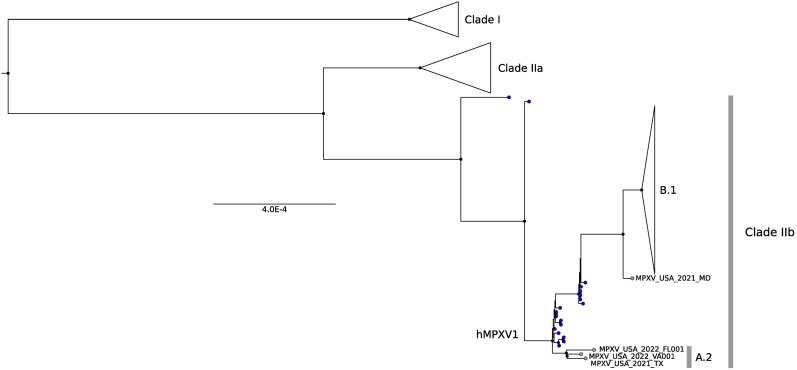
In the last five years, the prevalence of monkeypox has been increasing both in the regions considered endemic for the disease (West and Central Africa) and worldwide. Indeed, in July 2022, the World Health Organization declared the ongoing global outbreak of monkeypox a public health emergency of international concern. The disease is caused by monkeypox virus (MPXV), a member of the Orthopoxvirus genus, which also includes variola virus (the causative agent of smallpox) and vaccinia virus (used in the smallpox eradication campaign). Here, we review aspects of MPXV genetic diversity and epidemiology, with an emphasis on its genome structure, host range, and relationship with other orthopoxviruses. We also summarize the most recent findings deriving from the sequencing of outbreak MPXV genomes, and we discuss the apparent changing of MPXV evolutionary trajectory, which is characterized by the accumulation of point mutations rather than by gene gains/losses. Whereas the availability of a vaccine, the relatively mild presentation of the disease, and its relatively low transmissibility speak in favor of an efficient control of the global outbreak, the wide host range of MPXV raises concerns about the possible establishment of novel reservoirs. We also call for the deployment of field surveys and genomic surveillance programs to identify and control the MPXV reservoirs in West and Central Africa.
Keywords: Genetic diversity; Genome features; Geographic structure; Monkeypox; Zoonotic transmission.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Adalja A., Inglesby T. A novel international monkeypox outbreak. Ann. Intern. Med. 2022;75:1175–1176. - PubMed
-
- Adler H., Gould S., Hine P., Snell L.B., Wong W., Houlihan C.F., Osborne J.C., Rampling T., Beadsworth M.B., Duncan C.J., Dunning J., Fletcher T.E., Hunter E.R., Jacobs M., Khoo S.H., Newsholme W., Porter D., Porter R.J., Ratcliffe L., Schmid M.L., Semple M.G., Tunbridge A.J., Wingfield T., Price N.M., NHS England High Consequence Infectious Diseases (Airborne) Network Clinical features and management of human monkeypox: a retrospective observational study in the UK. Lancet Infect. Dis. 2022;22:1153–1162. - PMC - PubMed
-
- Alcamí A. Was smallpox a widespread mild disease? Science. 2020;369:376–377. - PubMed
-
- Alcamí A., Smith G.L. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 1992;71:153–167. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
