

Targeting replication stress in cancer therapy
- PMID: 36202931
- PMCID: PMC11132912
- DOI: 10.1038/s41573-022-00558-5
Targeting replication stress in cancer therapy
Abstract
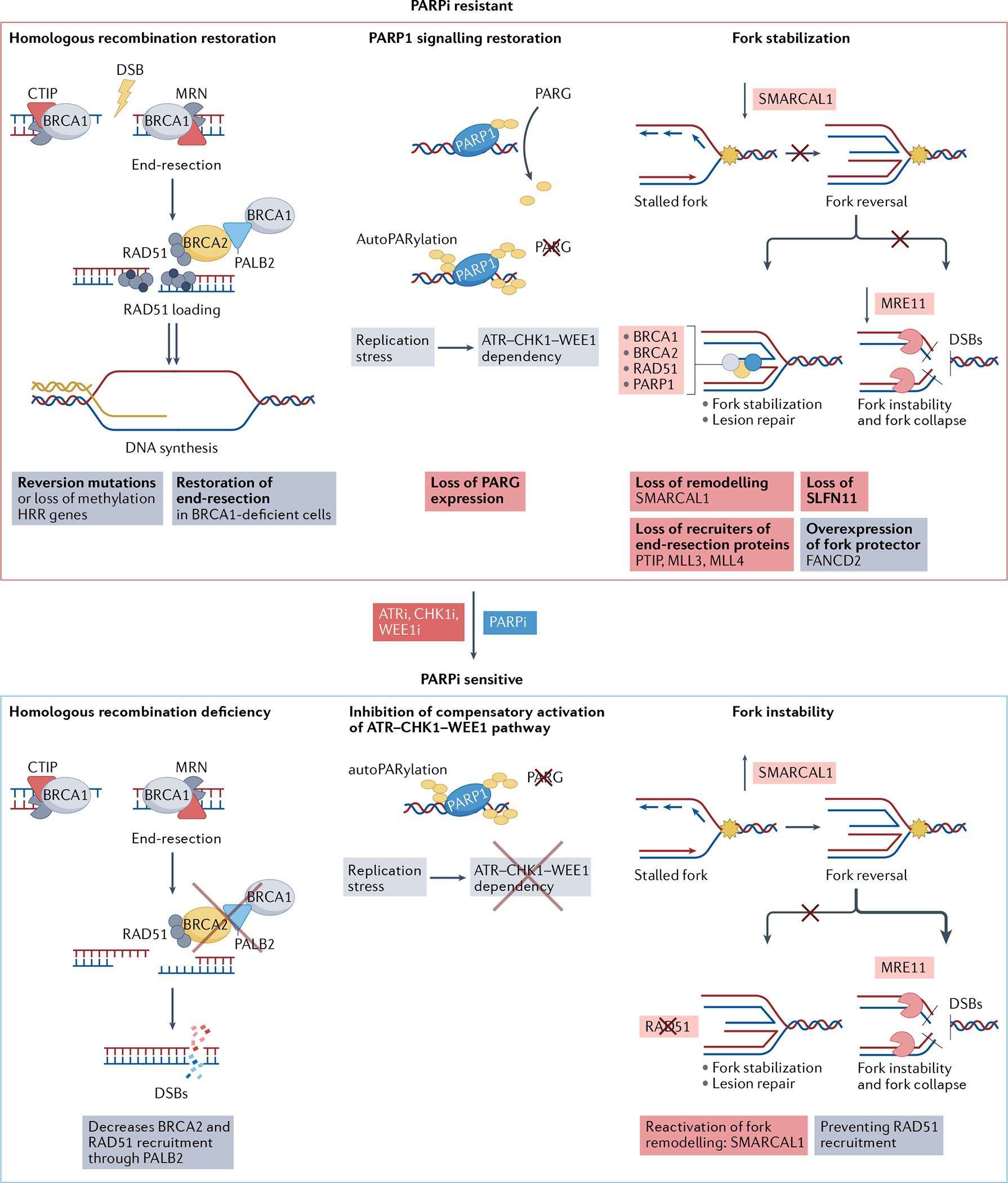
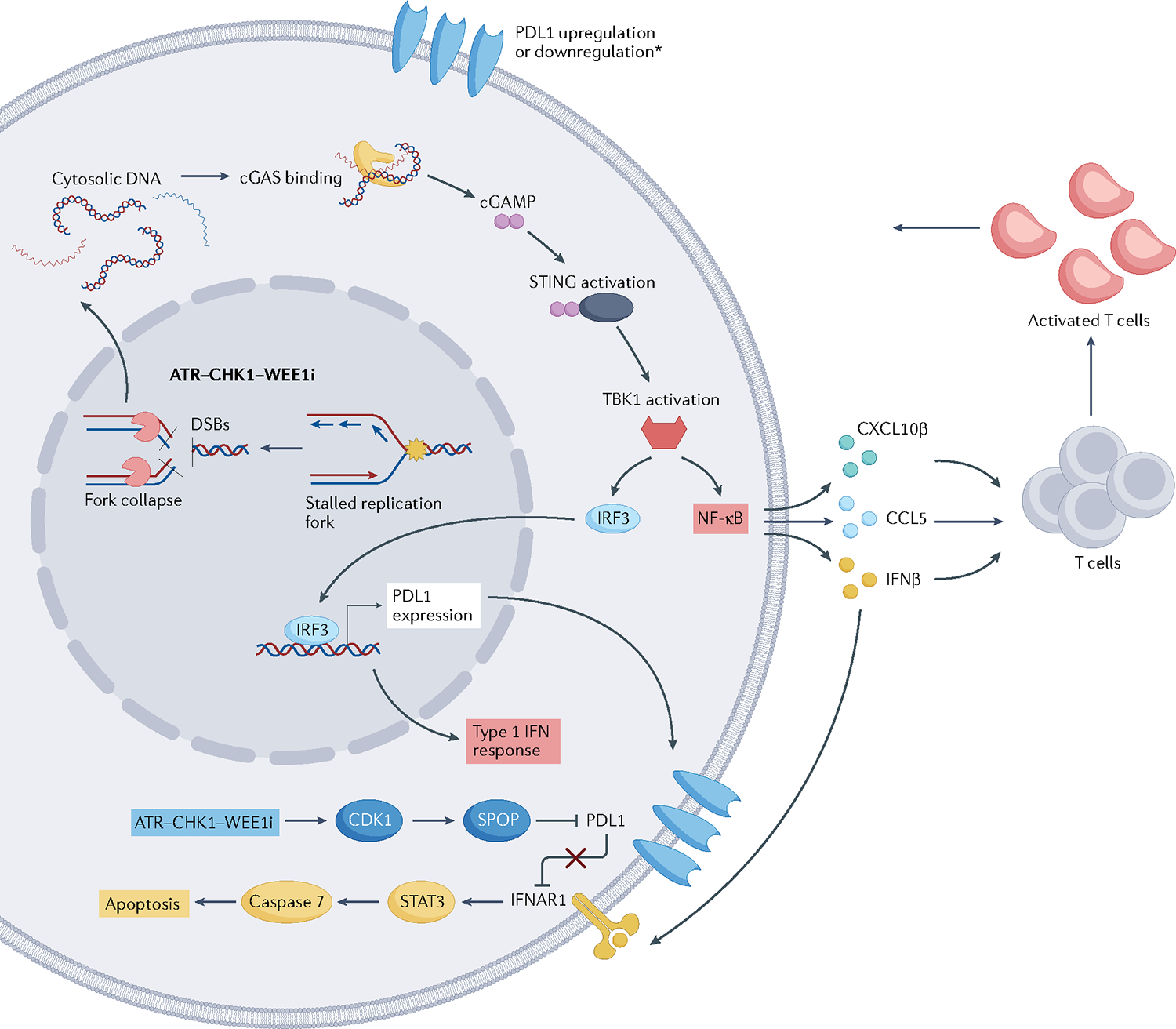
Replication stress is a major cause of genomic instability and a crucial vulnerability of cancer cells. This vulnerability can be therapeutically targeted by inhibiting kinases that coordinate the DNA damage response with cell cycle control, including ATR, CHK1, WEE1 and MYT1 checkpoint kinases. In addition, inhibiting the DNA damage response releases DNA fragments into the cytoplasm, eliciting an innate immune response. Therefore, several ATR, CHK1, WEE1 and MYT1 inhibitors are undergoing clinical evaluation as monotherapies or in combination with chemotherapy, poly[ADP-ribose]polymerase (PARP) inhibitors, or immune checkpoint inhibitors to capitalize on high replication stress, overcome therapeutic resistance and promote effective antitumour immunity. Here, we review current and emerging approaches for targeting replication stress in cancer, from preclinical and biomarker development to clinical trial evaluation.
© 2022. Springer Nature Limited.
Conflict of interest statement
Competing interests
G.I.S. is a consultant/advisory board member for Lilly, Sierra Oncology, Merck-EMD Serono, Pfizer, Astex, Almac, Roche, Bicycle Therapeutics, Fusion Pharmaceuticals, G1 Therapeutics, Bayer, Ip-sen, Cybrexa Therapeutics, Angiex, Daiichi Sankyo and Seattle Genetics, and reports receipt of commercial research grants from Lilly, Sierra Oncology, Merck-EMD Serono and Merck & Co. P.A.K. reports participation in advisory boards from GlaxoSmithKline/Tesaro, Merck, AstraZeneca and Bayer. A.D.D. is a consultant and/or advisory board member for AstraZeneca, Bayer AG, Cedilla Therapeutics, Celgene, Cyteir Therapeutics, Epizyme, GalaxoSmithKline, Ideaya, Impact Therapeutics, LAV Global Management Company Limited. D.C. and A.A.B.A.C. declare no competing interests.
Figures
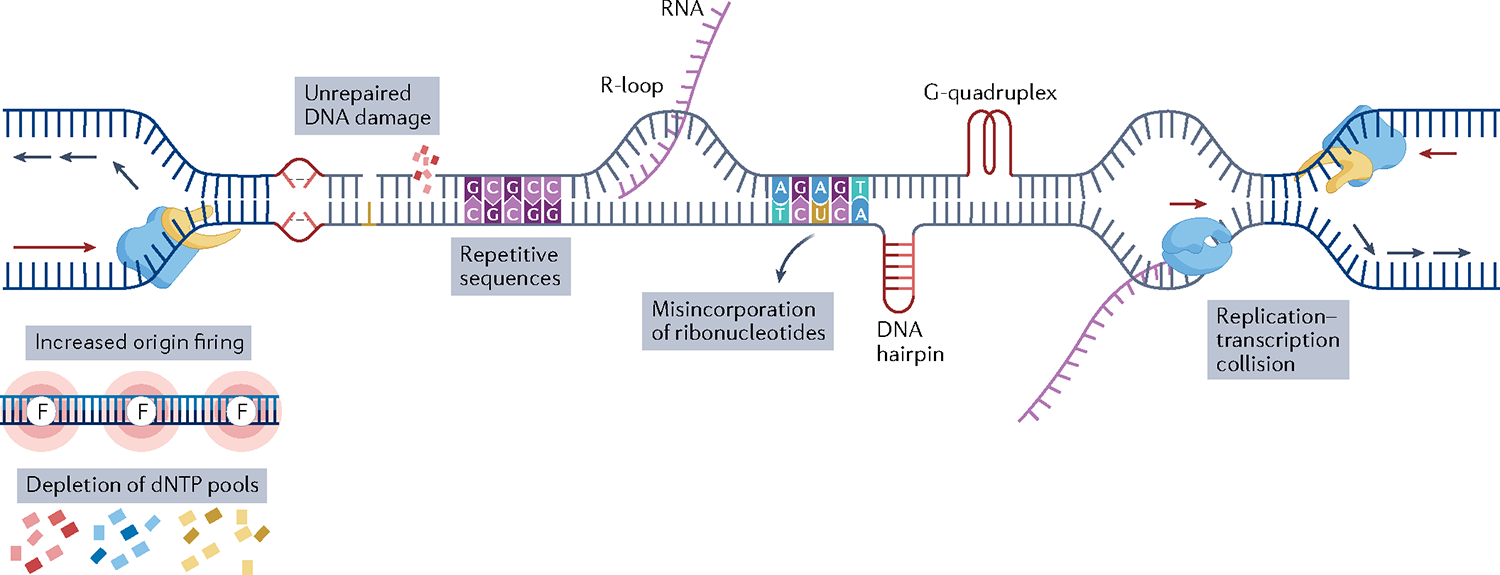
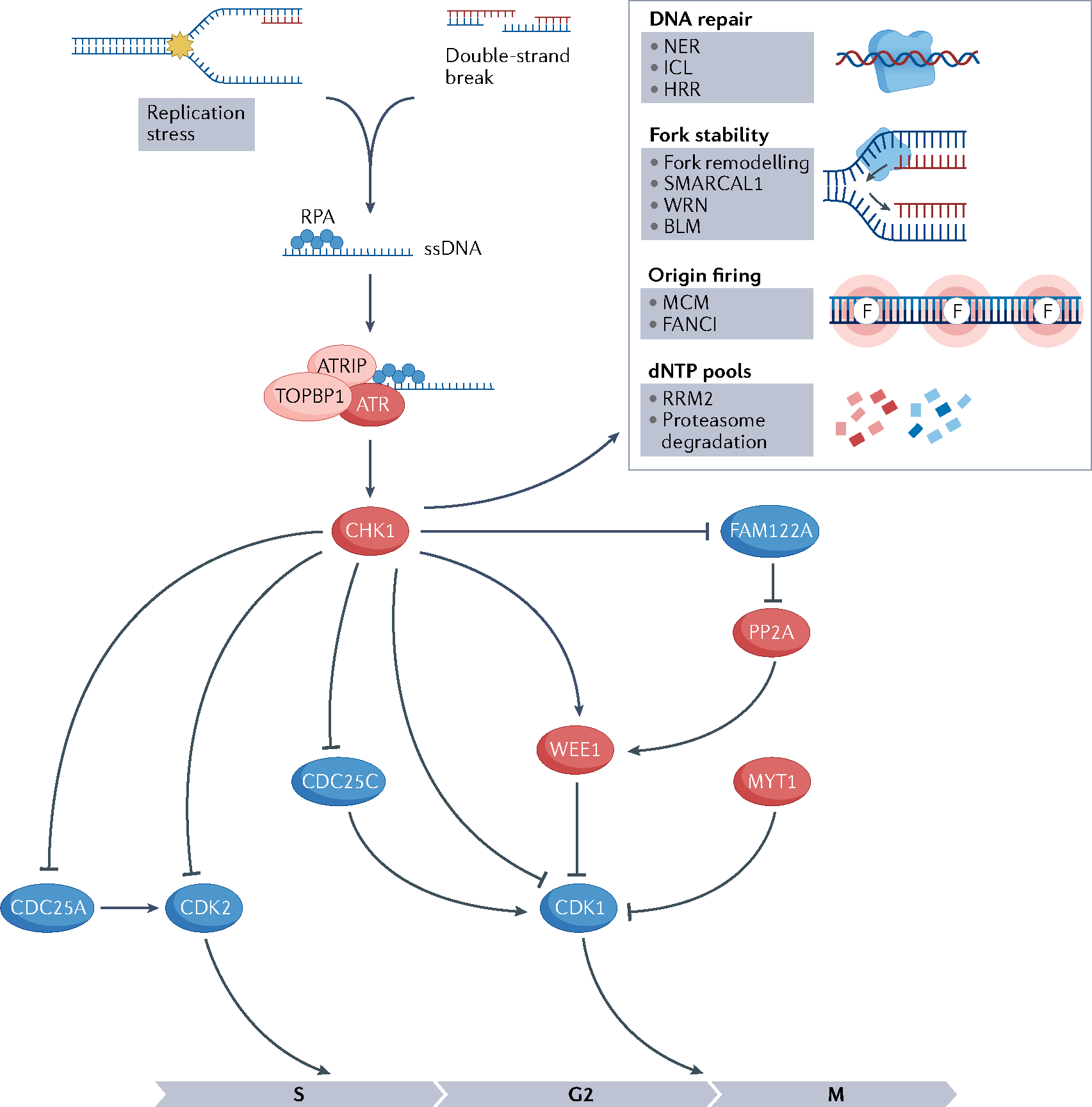
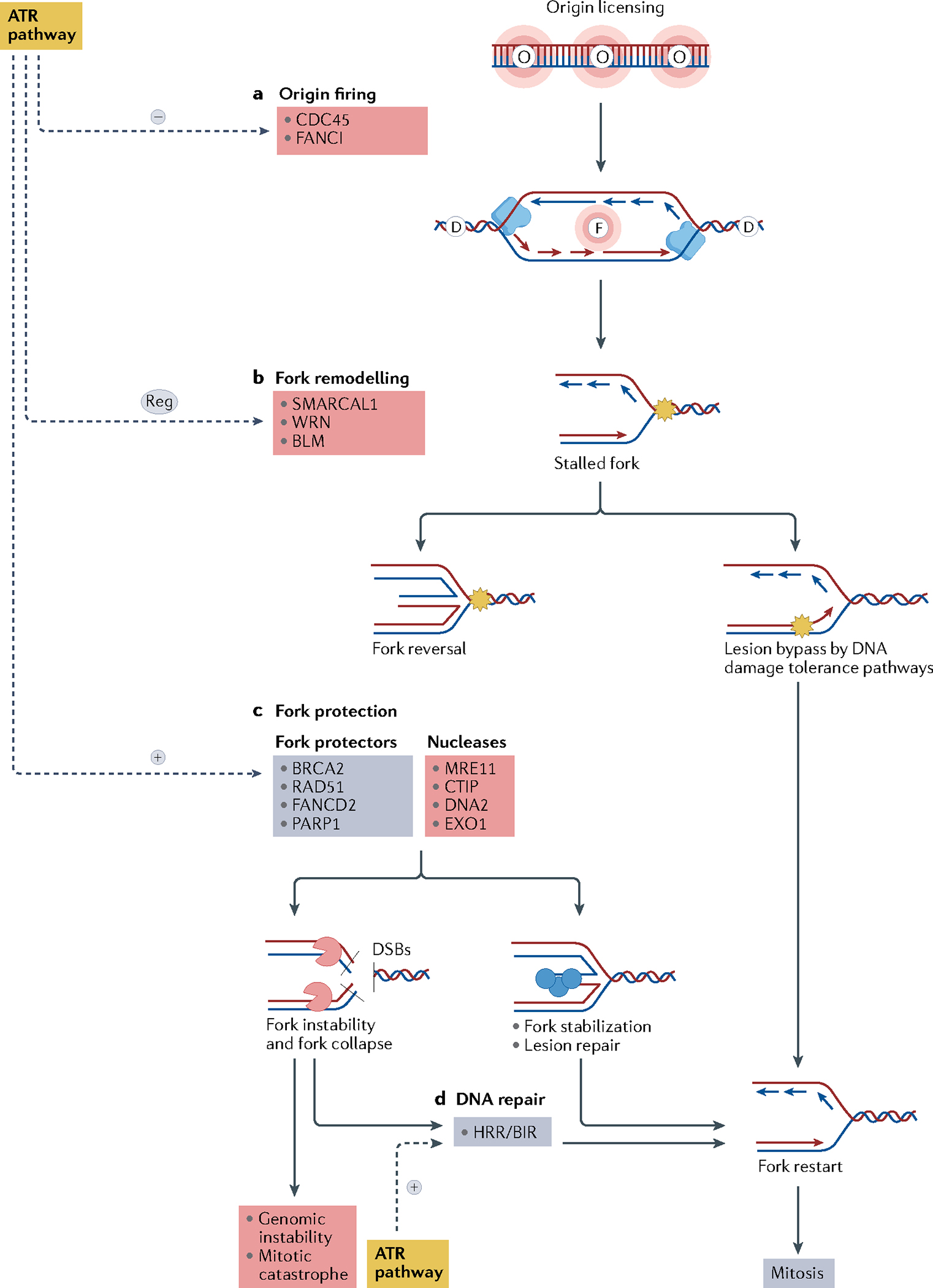
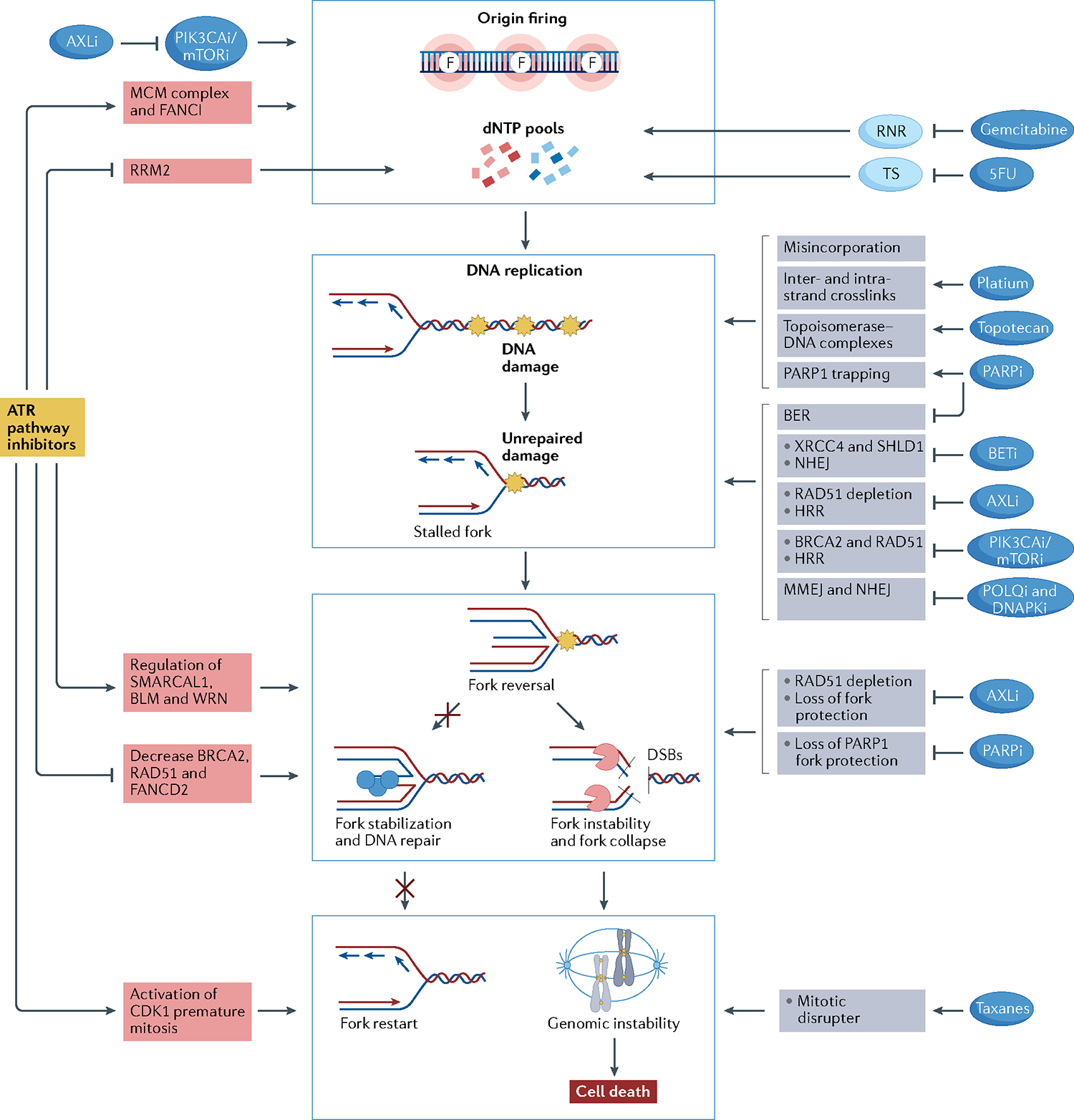
References
-
- Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). - PubMed
-
- Gaillard H, García-Muse T & Aguilera A Replication stress and cancer. Nat. Rev. Cancer 15, 276–280 (2015). - PubMed
-
- Halazonetis TD, Gorgoulis VG & Bartek J An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
