

A PKA inhibitor motif within SMOOTHENED controls Hedgehog signal transduction
- PMID: 36202993
- PMCID: PMC9696579
- DOI: 10.1038/s41594-022-00838-z
A PKA inhibitor motif within SMOOTHENED controls Hedgehog signal transduction
Abstract
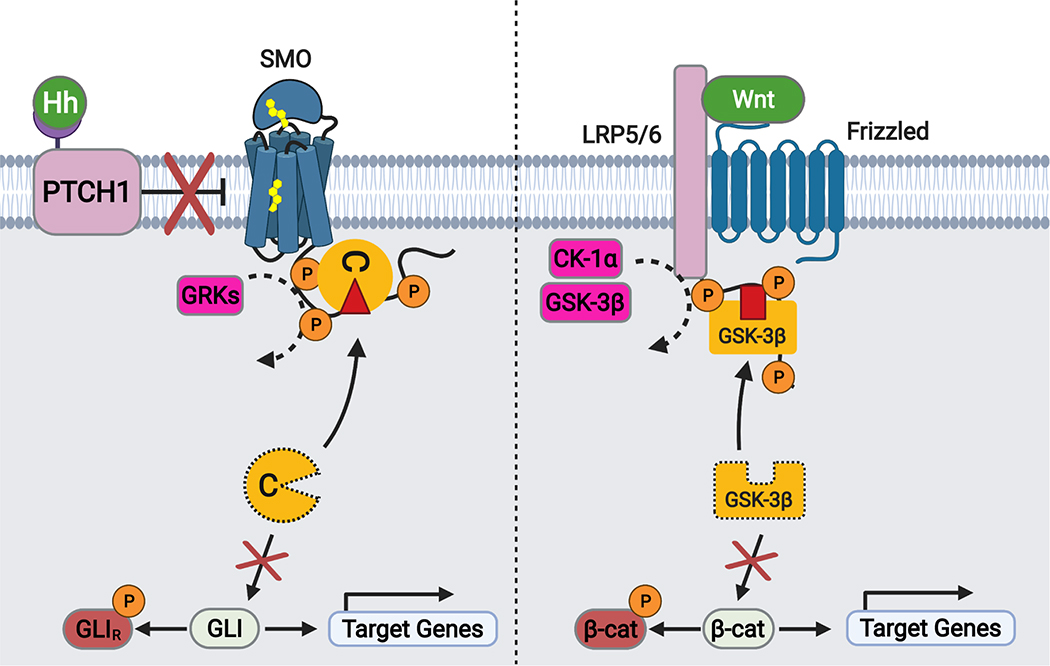
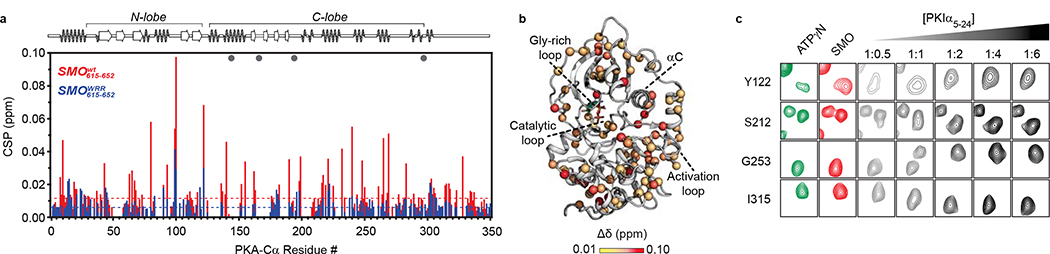
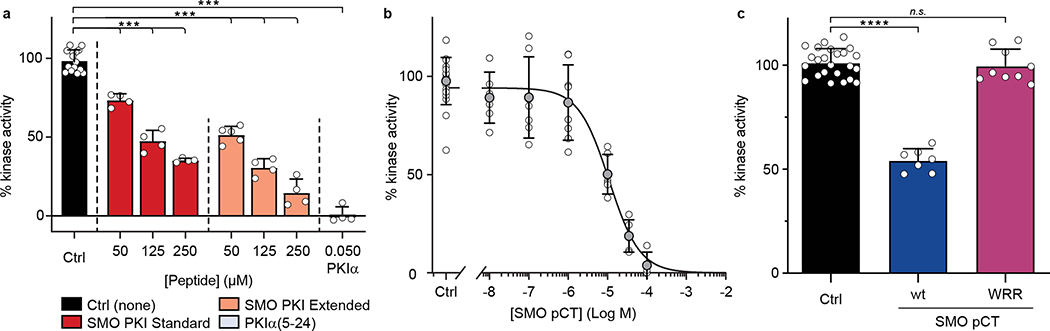
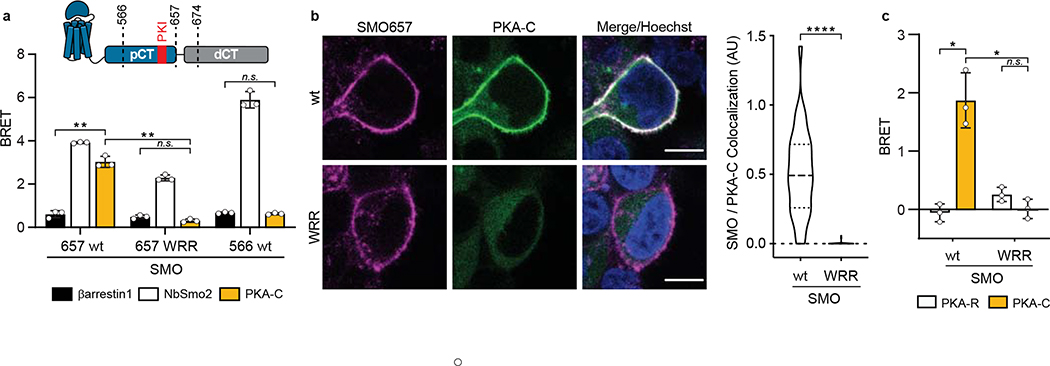
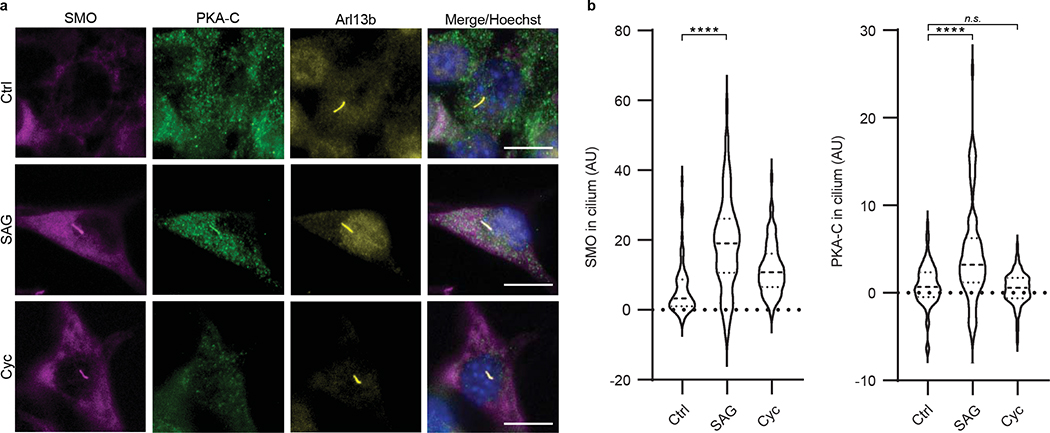
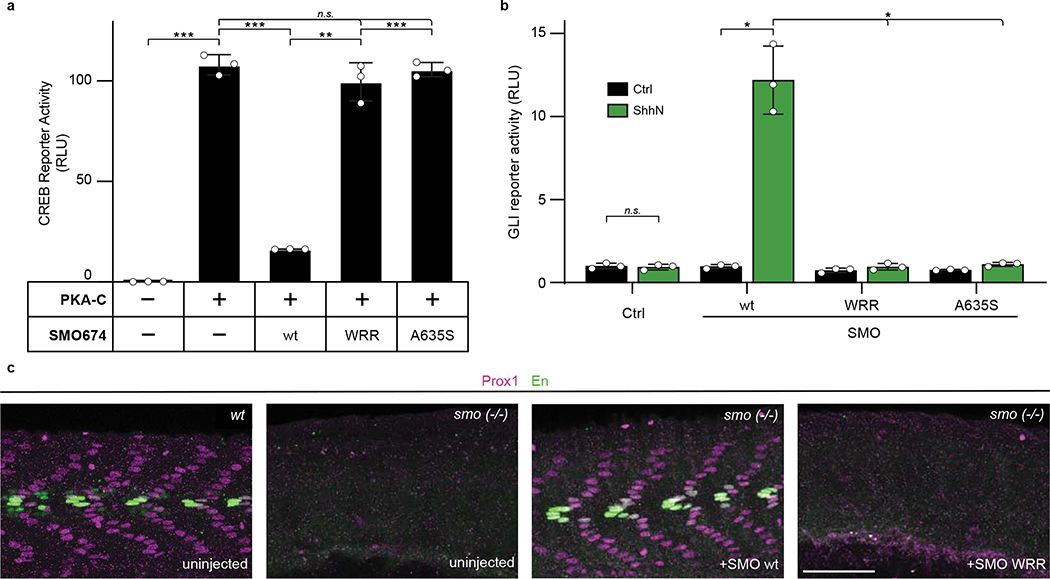
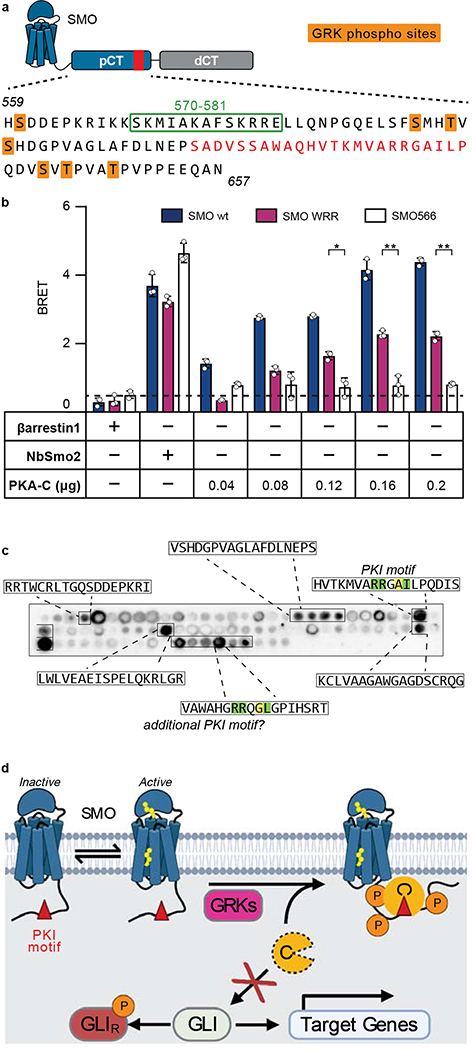
The Hedgehog (Hh) cascade is central to development, tissue homeostasis and cancer. A pivotal step in Hh signal transduction is the activation of glioma-associated (GLI) transcription factors by the atypical G protein-coupled receptor (GPCR) SMOOTHENED (SMO). How SMO activates GLI remains unclear. Here we show that SMO uses a decoy substrate sequence to physically block the active site of the cAMP-dependent protein kinase (PKA) catalytic subunit (PKA-C) and extinguish its enzymatic activity. As a result, GLI is released from phosphorylation-induced inhibition. Using a combination of in vitro, cellular and organismal models, we demonstrate that interfering with SMO-PKA pseudosubstrate interactions prevents Hh signal transduction. The mechanism uncovered echoes one used by the Wnt cascade, revealing an unexpected similarity in how these two essential developmental and cancer pathways signal intracellularly. More broadly, our findings define a mode of GPCR-PKA communication that may be harnessed by a range of membrane receptors and kinases.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS:
The authors declare no competing financial interests.
Figures

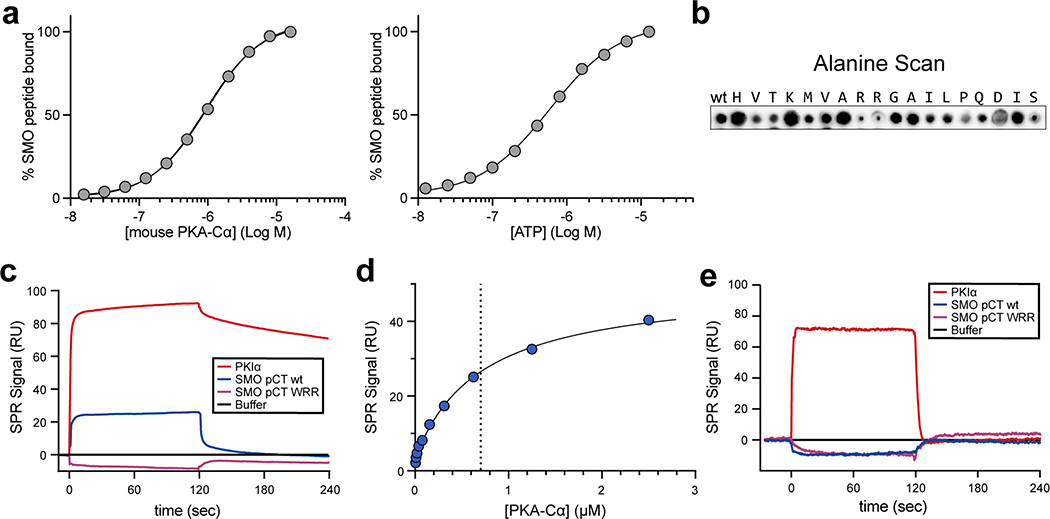
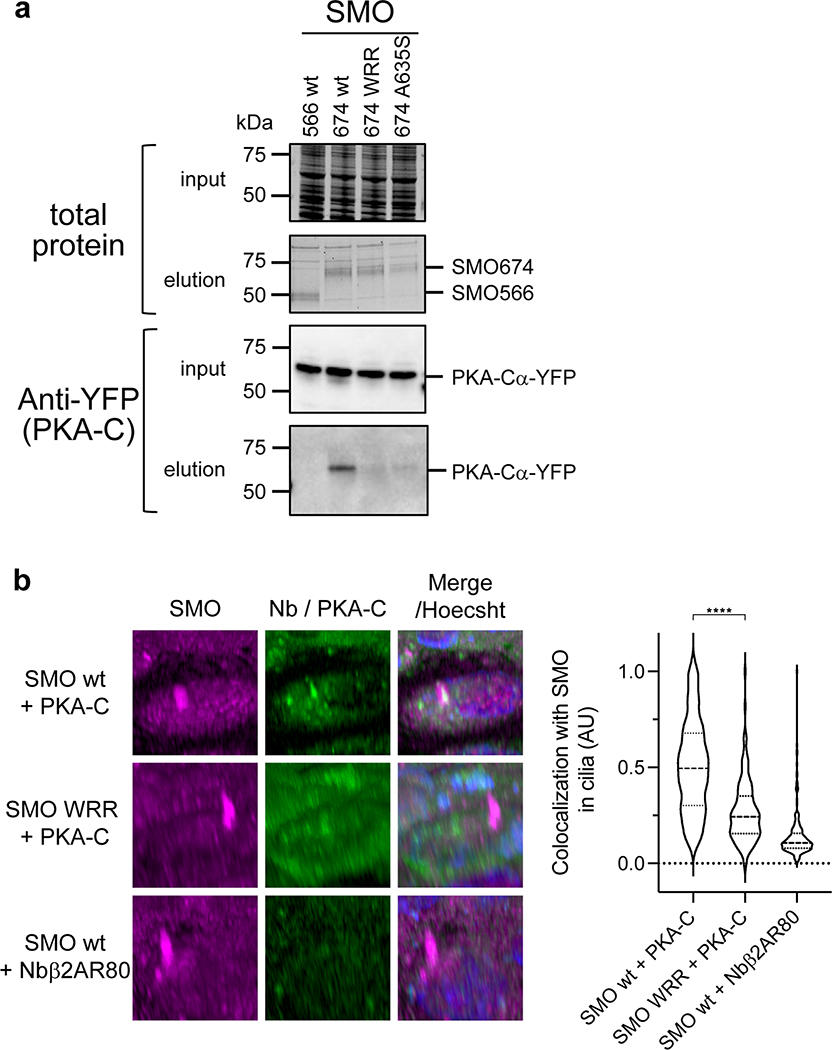
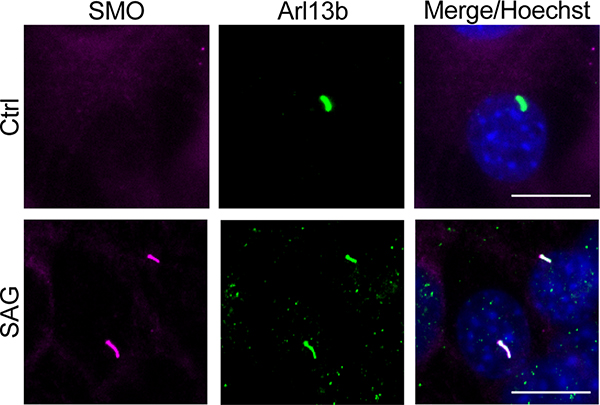
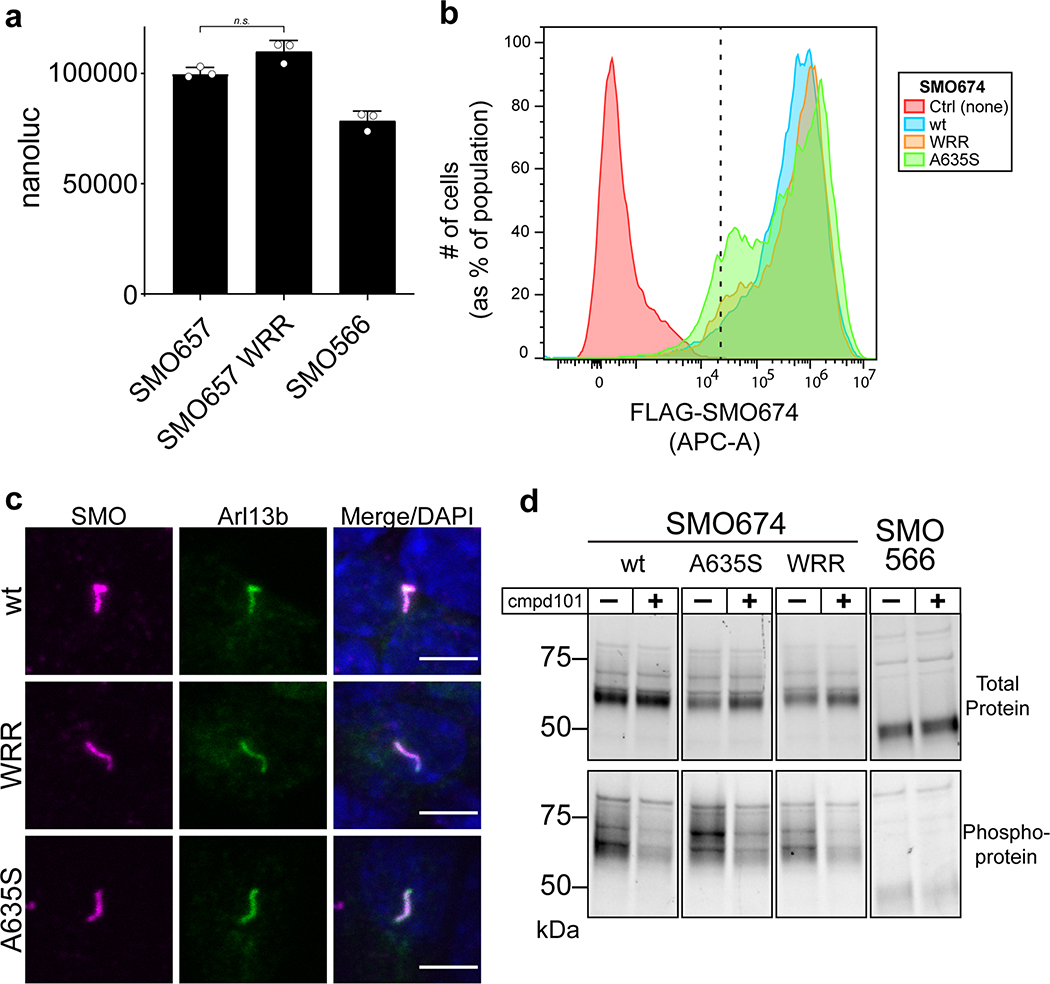
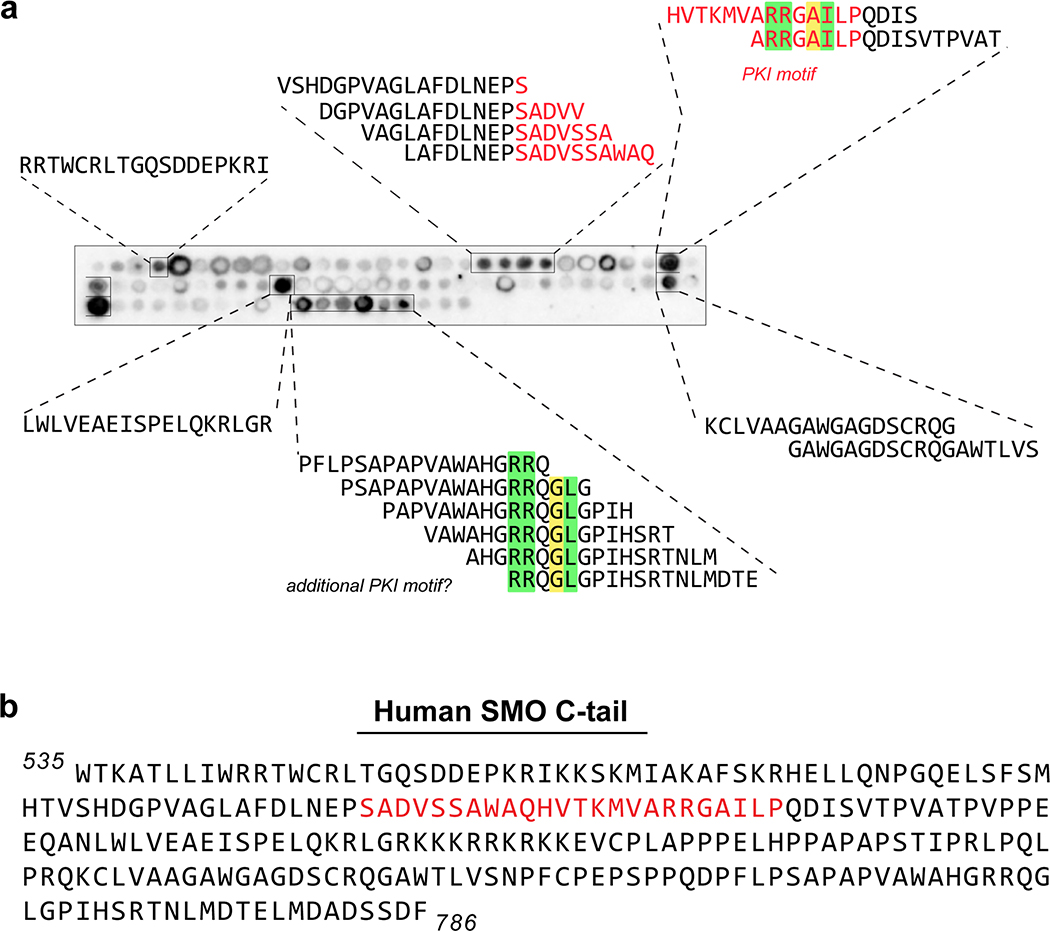

References
-
- Briscoe J & Therond PP The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14, 416–29 (2013). - PubMed
-
- Ingham PW & McMahon AP Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15, 3059–87 (2001). - PubMed
-
- Ingham PW, Nakano Y & Seger C Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet 12, 393–406 (2011). - PubMed
-
- Muenke M & Beachy PA Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev 10, 262–9 (2000). - PubMed
METHODS-ONLY REFERENCES:
-
- Zimmermann B, Schweinsberg S, Drewianka S & Herberg FW Effect of metal ions on high-affinity binding of pseudosubstrate inhibitors to PKA. Biochem J 413, 93–101 (2008). - PubMed
-
- Olsen SR & Uhler MD Affinity purification of the C alpha and C beta isoforms of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem 264, 18662–6 (1989). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
