

Functional inference of gene regulation using single-cell multi-omics
- PMID: 36204155
- PMCID: PMC9534481
- DOI: 10.1016/j.xgen.2022.100166
Functional inference of gene regulation using single-cell multi-omics
Abstract
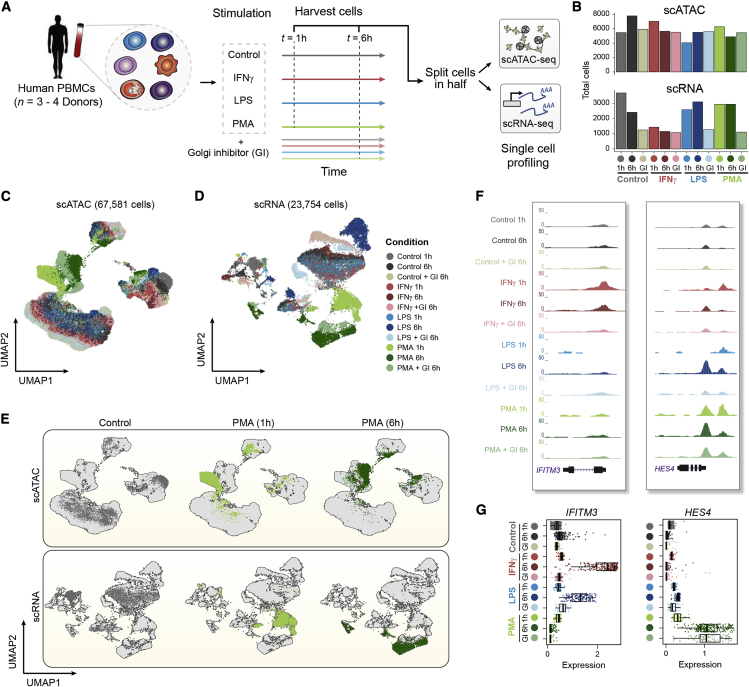
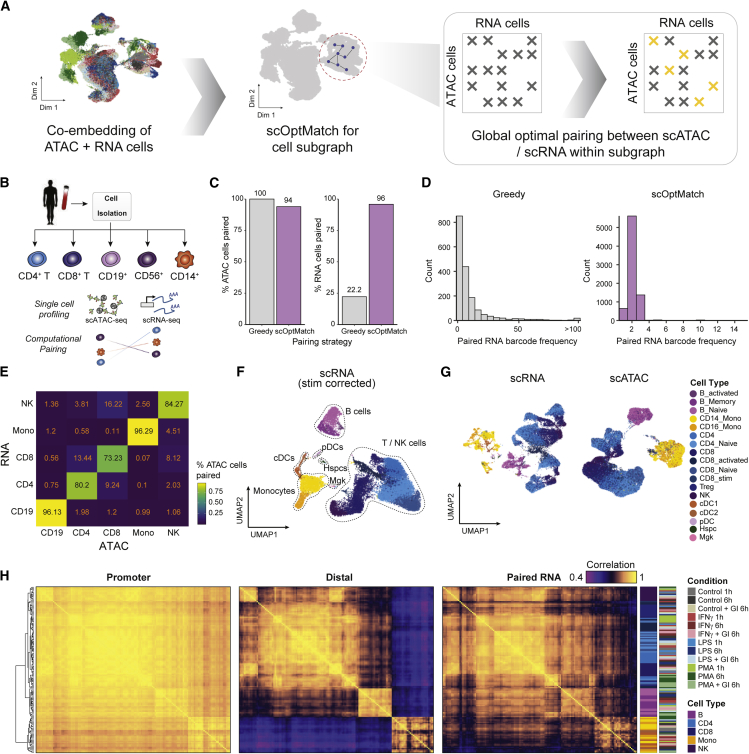
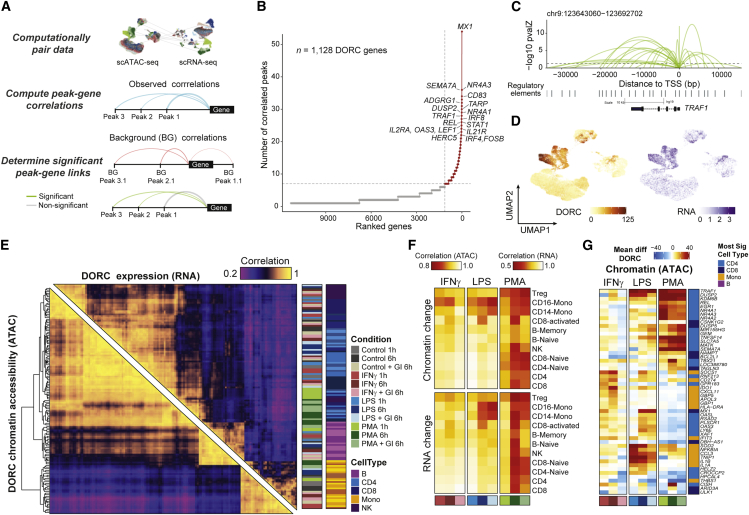
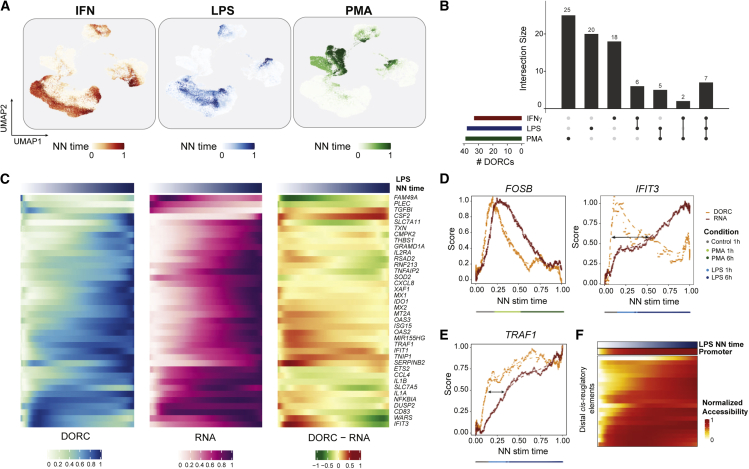
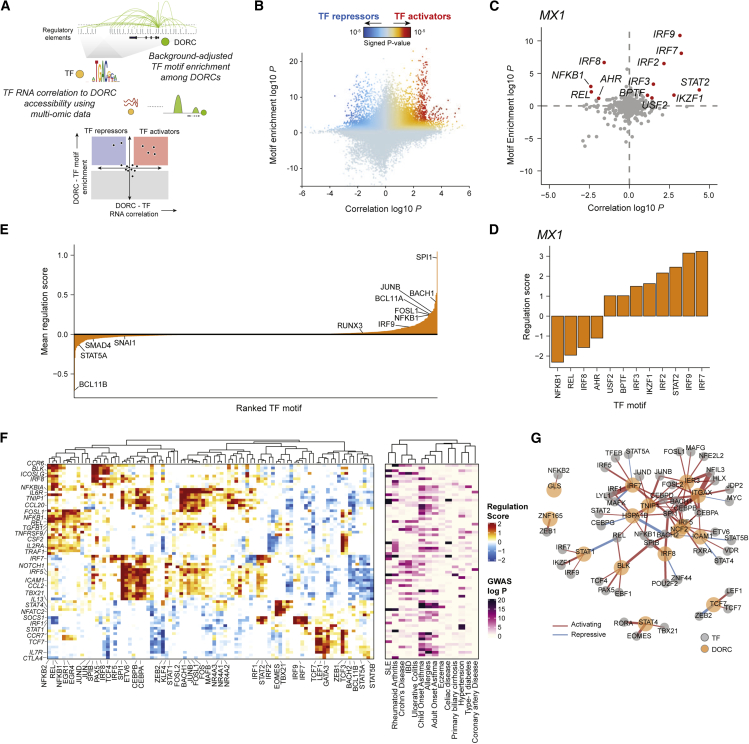
Cells require coordinated control over gene expression when responding to environmental stimuli. Here we apply scATAC-seq and single-cell RNA sequencing (scRNA-seq) in resting and stimulated human blood cells. Collectively, we generate ~91,000 single-cell profiles, allowing us to probe the cis-regulatory landscape of the immunological response across cell types, stimuli, and time. Advancing tools to integrate multi-omics data, we develop functional inference of gene regulation (FigR), a framework to computationally pair scA-TAC-seq with scRNA-seq cells, connect distal cis-regulatory elements to genes, and infer gene-regulatory networks (GRNs) to identify candidate transcription factor (TF) regulators. Utilizing these paired multi-omics data, we define domains of regulatory chromatin (DORCs) of immune stimulation and find that cells alter chromatin accessibility and gene expression at timescales of minutes. Construction of the stimulation GRN elucidates TF activity at disease-associated DORCs. Overall, FigR enables elucidation of regulatory interactions across single-cell data, providing new opportunities to understand the function of cells within tissues.
Conflict of interest statement
DECLARATION OF INTERESTS J.D.B. holds patents related to ATAC-seq and scATAC-seq and serves on the scientific advisory boards of CAMP4 Therapeutics, seqWell, and CelSee. J.G.C., Z.D.B., A.S.K., and R.L. are employees of Bio-Rad.
Figures
References
-
- Klemm S.L., Shipony Z., Greenleaf W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019;20:207–220. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
