

Targeting telomerase reverse transcriptase with the covalent inhibitor NU-1 confers immunogenic radiation sensitization
- PMID: 36206753
- PMCID: PMC9588800
- DOI: 10.1016/j.chembiol.2022.09.002
Targeting telomerase reverse transcriptase with the covalent inhibitor NU-1 confers immunogenic radiation sensitization
Abstract
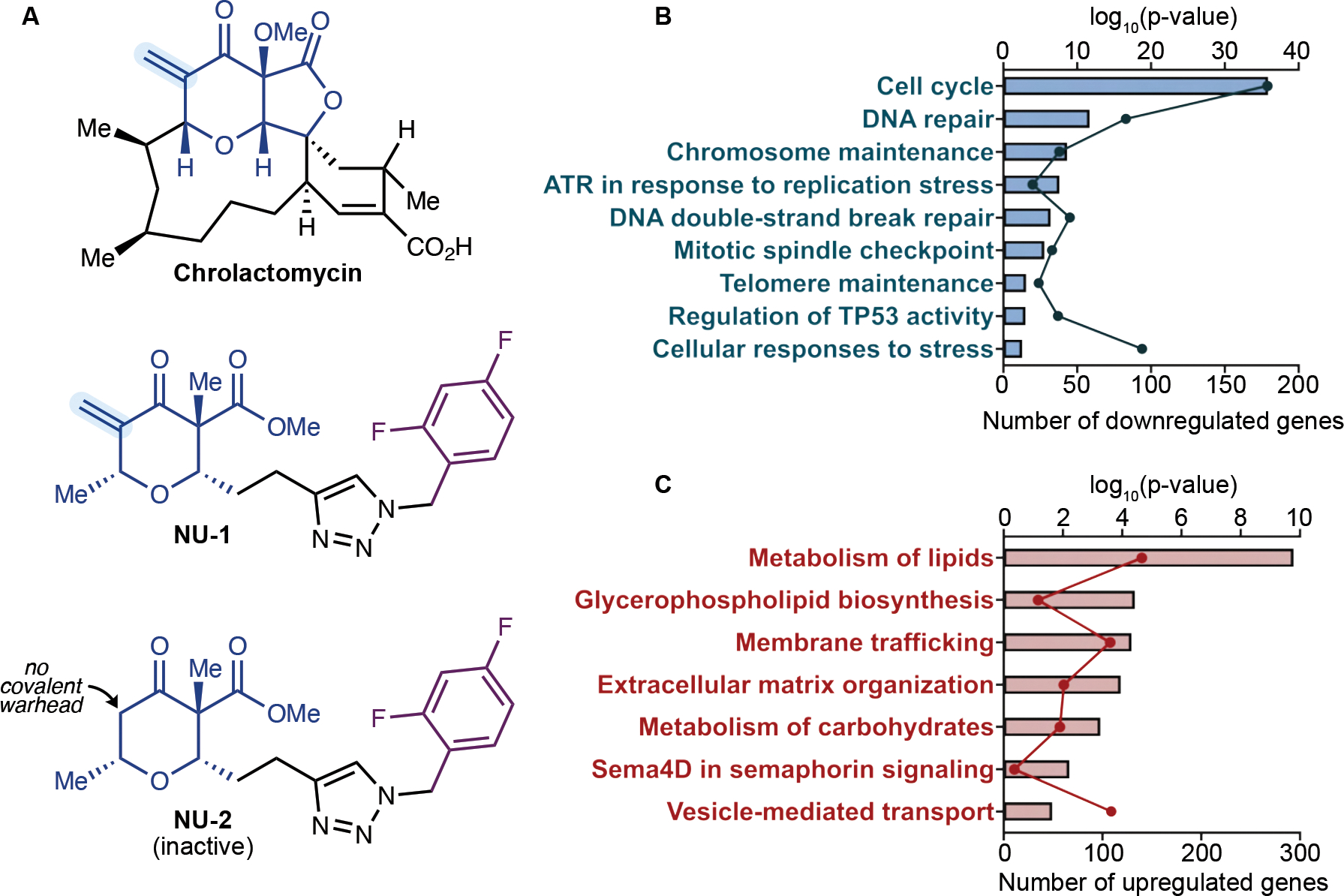
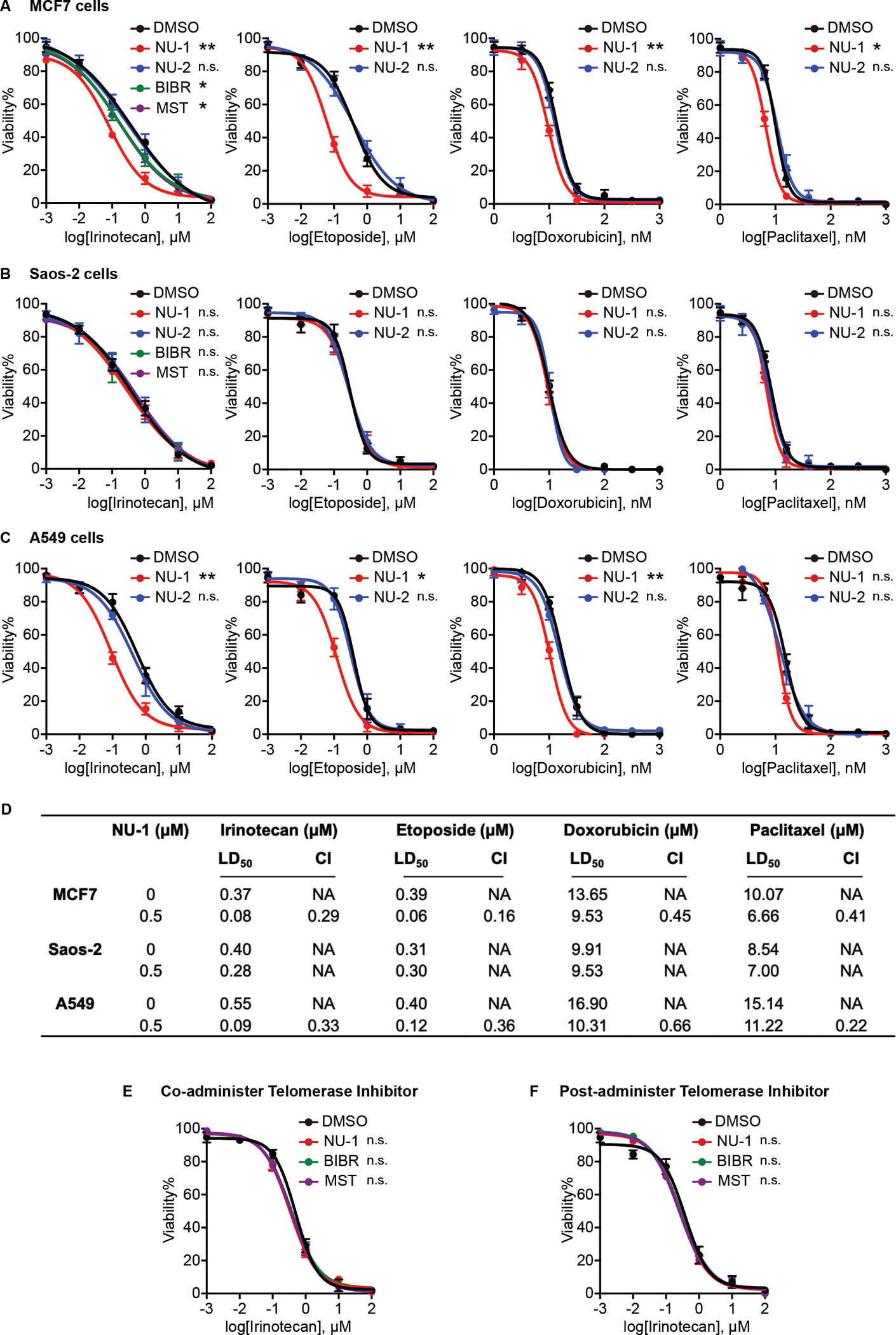
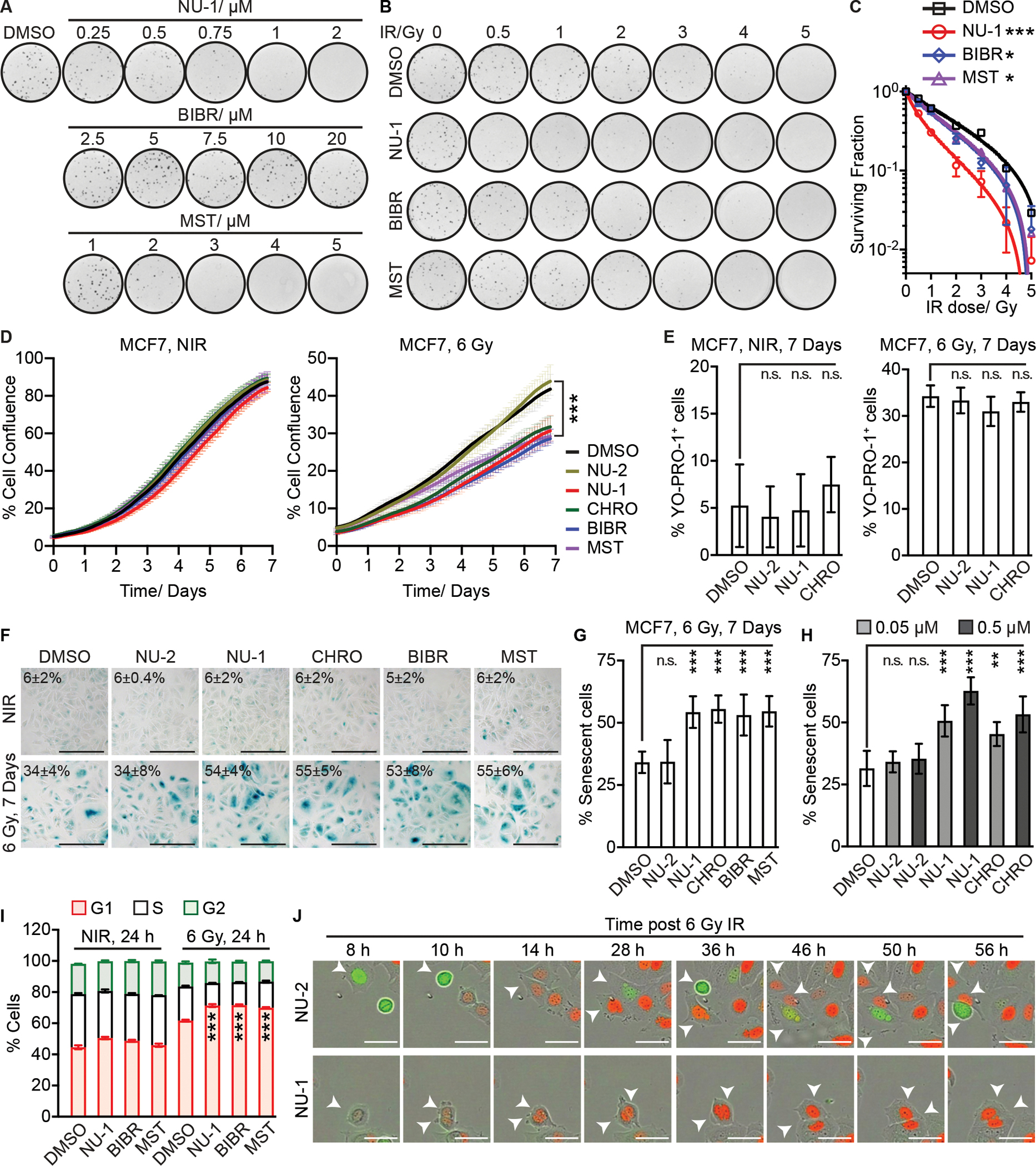
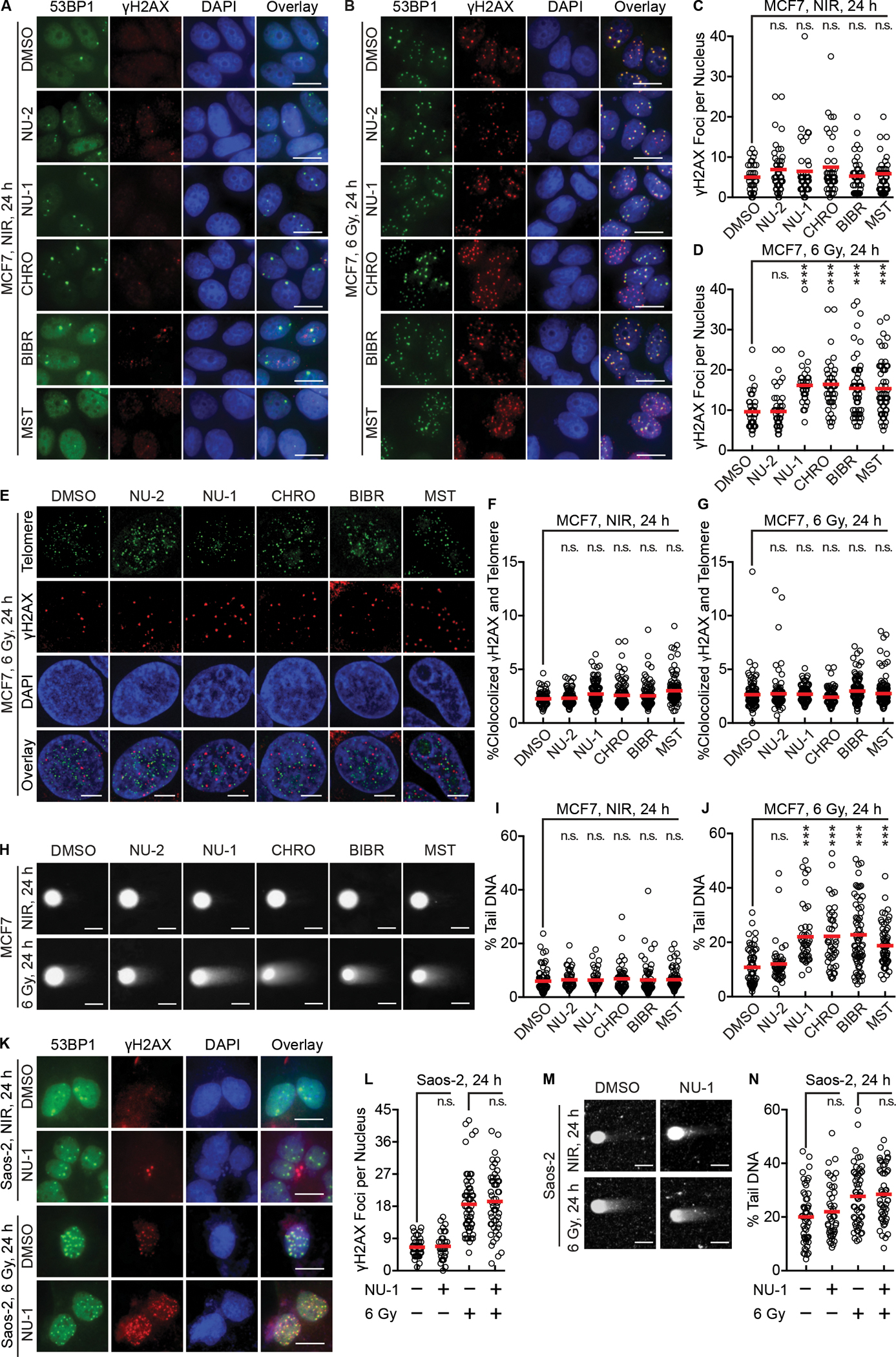
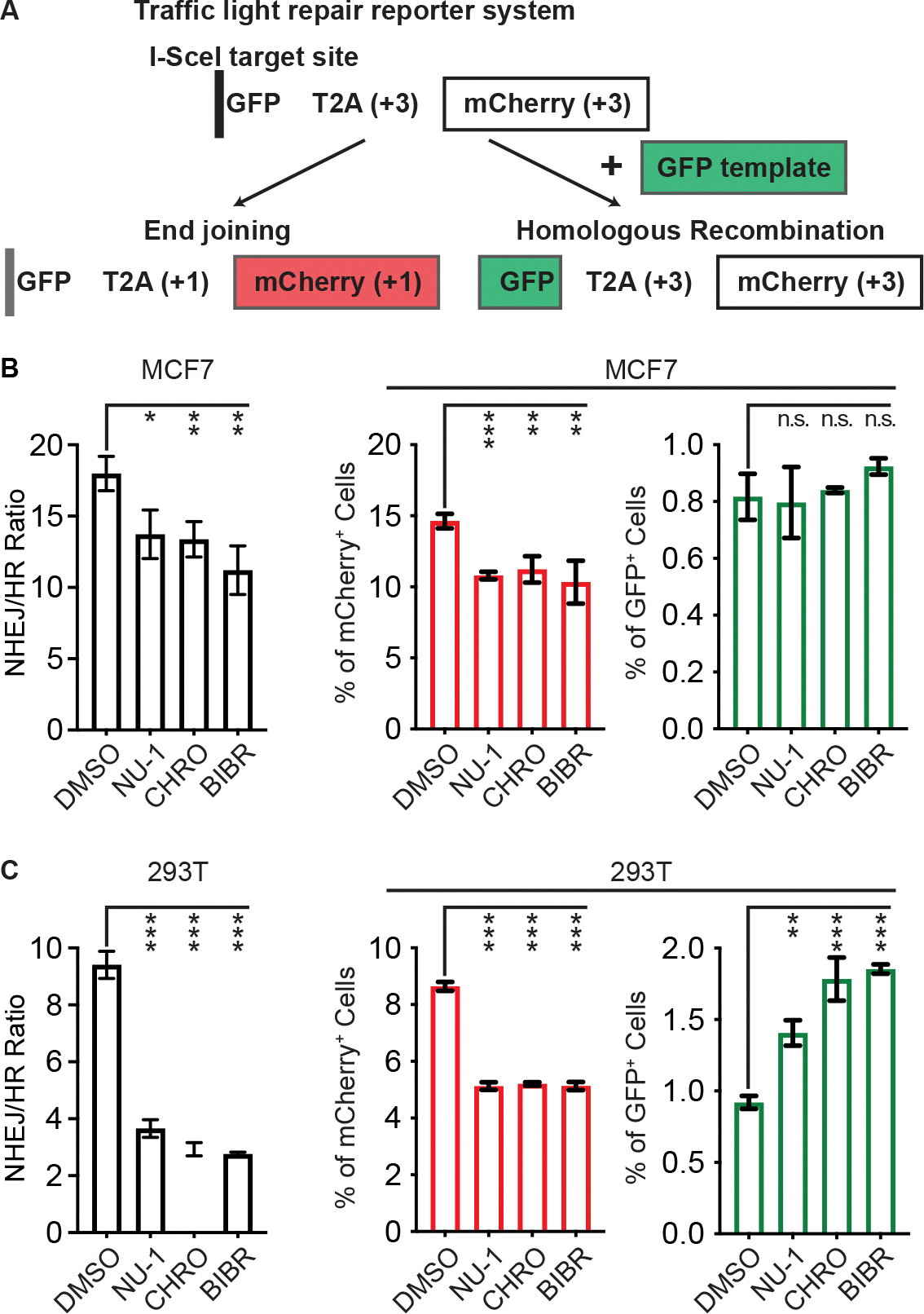
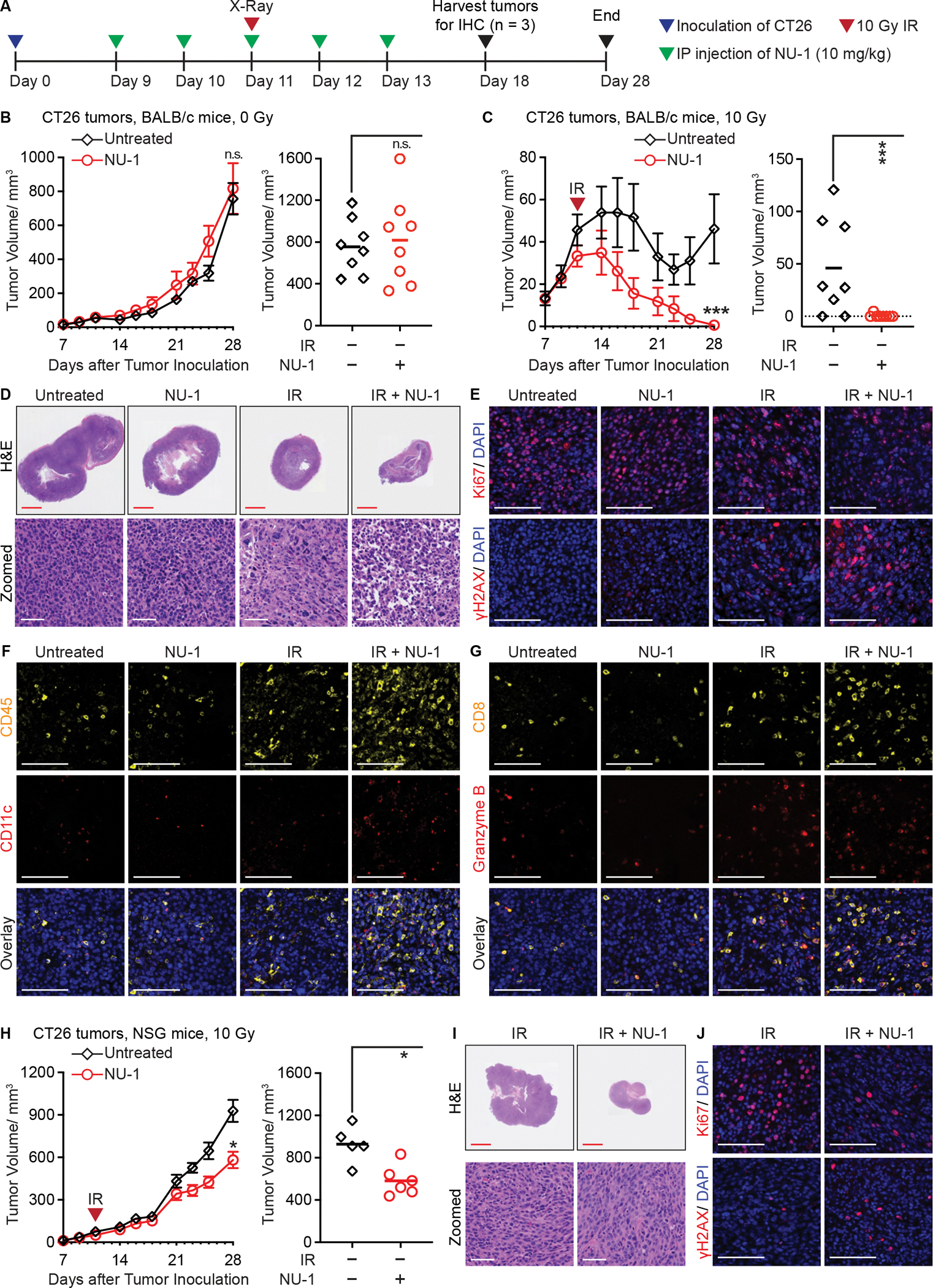
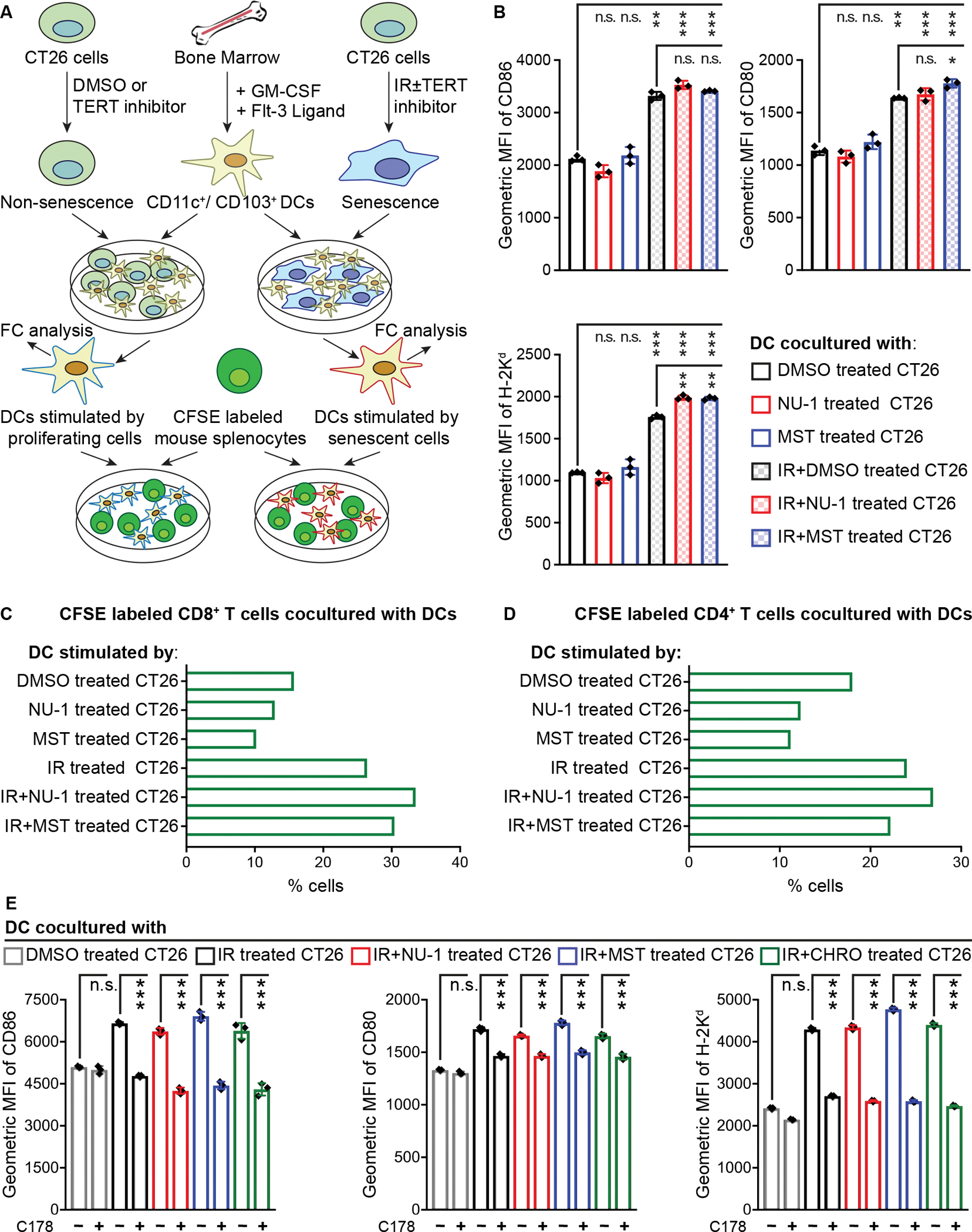
Beyond synthesizing telomere repeats, the telomerase reverse transcriptase (TERT) also serves multiple other roles supporting cancer growth. Blocking telomerase to drive telomere erosion appears impractical, but TERT's non-canonical activities have yet to be fully explored as cancer targets. Here, we used an irreversible TERT inhibitor, NU-1, to examine impacts on resistance to conventional cancer therapies. In vitro, inhibiting TERT sensitized cells to chemotherapy and radiation. NU-1 delayed repair of double-strand breaks, resulting in persistent DNA damage signaling and cellular senescence. Although NU-1 alone did not impact growth of syngeneic CT26 tumors in BALB/c mice, it dramatically enhanced the effects of radiation, leading to immune-dependent tumor elimination. Tumors displayed persistent DNA damage, suppressed proliferation, and increased activated immune infiltrate. Our studies confirm TERT's role in limiting genotoxic effects of conventional therapy but also implicate TERT as a determinant of immune evasion and therapy resistance.
Keywords: DNA damage response; TERT; anti-tumor immunity; combination therapy; radiation; senescence.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests Y.L., R.C.B., G.B.F., K.A.S., and S.J.K. are co-inventors of intellectual property owned by Northwestern University and the University of Chicago and co-founders and/or co-owners of Riptide Therapeutics, a spin-out from Northwestern University and the University of Chicago, related to applications of chrolactomycin analog TERT inhibitors in cancer therapeutics. R.C.B. is an employee of Corteva Agriscience. S.J.K. is a co-founder of OncoSenescence.
Figures
References
-
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, and Saretzki G (2008). Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. Journal of cell science 121, 1046–1053. - PubMed
-
- Akıncılar SC, Low KC, Liu CY, Yan TD, Oji A, Ikawa M, Li S, and Tergaonkar V (2015). Quantitative assessment of telomerase components in cancer cell lines. FEBS letters 589, 974–984. - PubMed
-
- Akiyama M, Yamada O, Kanda N, Akita S, Kawano T, Ohno T, Mizoguchi H, Eto Y, Anderson KC, and Yamada H (2002). Telomerase overexpression in K562 leukemia cells protects against apoptosis by serum deprivation and double-stranded DNA break inducing agents, but not against DNA synthesis inhibitors. Cancer Lett 178, 187–197. 10.1016/s0304-3835(01)00838-2. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
