

Axon Biology in ALS: Mechanisms of Axon Degeneration and Prospects for Therapy
- PMID: 36207571
- PMCID: PMC9587191
- DOI: 10.1007/s13311-022-01297-6
Axon Biology in ALS: Mechanisms of Axon Degeneration and Prospects for Therapy
Abstract
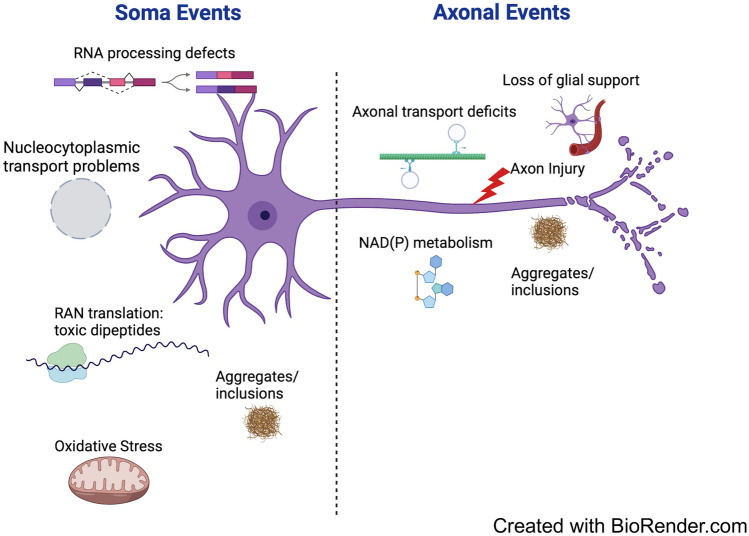
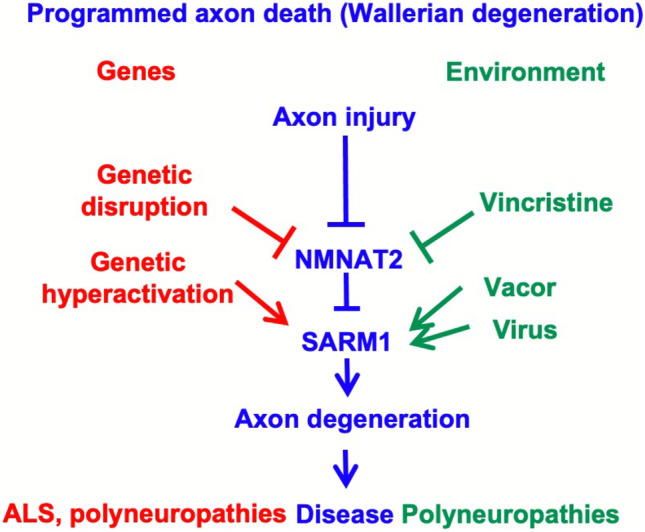
This review addresses the longstanding debate over whether amyotrophic lateral sclerosis (ALS) is a 'dying back' or 'dying forward' disorder in the light of new gene identifications and the increased understanding of mechanisms of action for previously identified ALS genes. While the topological pattern of pathology in animal models, and more anecdotally in patients is indeed 'dying back', this review discusses how this fits with the fact that many of the major initiating events are thought to occur within the soma. It also discusses how widely varying ALS risk factors, including some impacting axons directly, may combine to drive a common pathway involving TAR DNA binding protein 43 (TDP-43) and neuromuscular junction (NMJ) denervation. The emerging association between sterile alpha and TIR motif-containing 1 (SARM1), a protein so far mostly associated with axon degeneration, and sporadic ALS is another major theme. The strengths and limitations of the current evidence supporting an association are considered, along with ways in which SARM1 could become activated in ALS. The final section addresses SARM1-based therapies along with the prospects for targeting other axonal steps in ALS pathogenesis.
Keywords: Axon degeneration; Axonal transport; NAD; NMNAT2; Programmed axon death; SARM1.
© 2022. The Author(s).
Figures
References
-
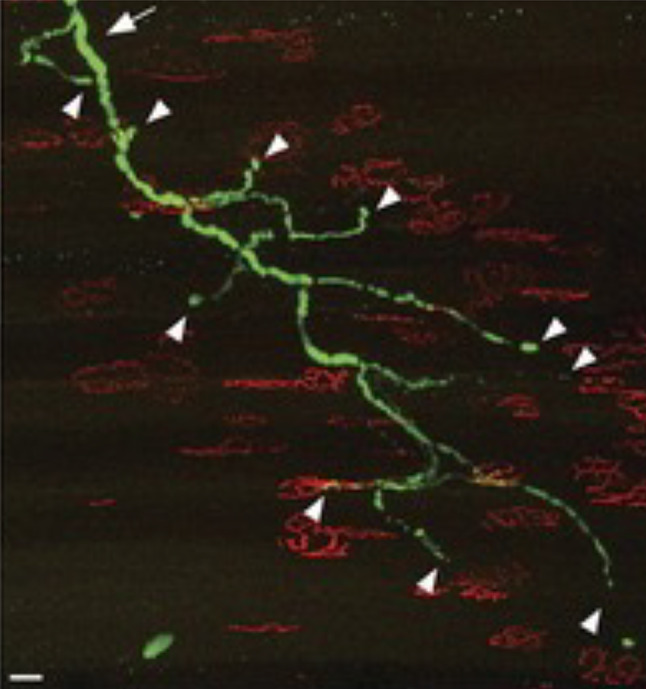
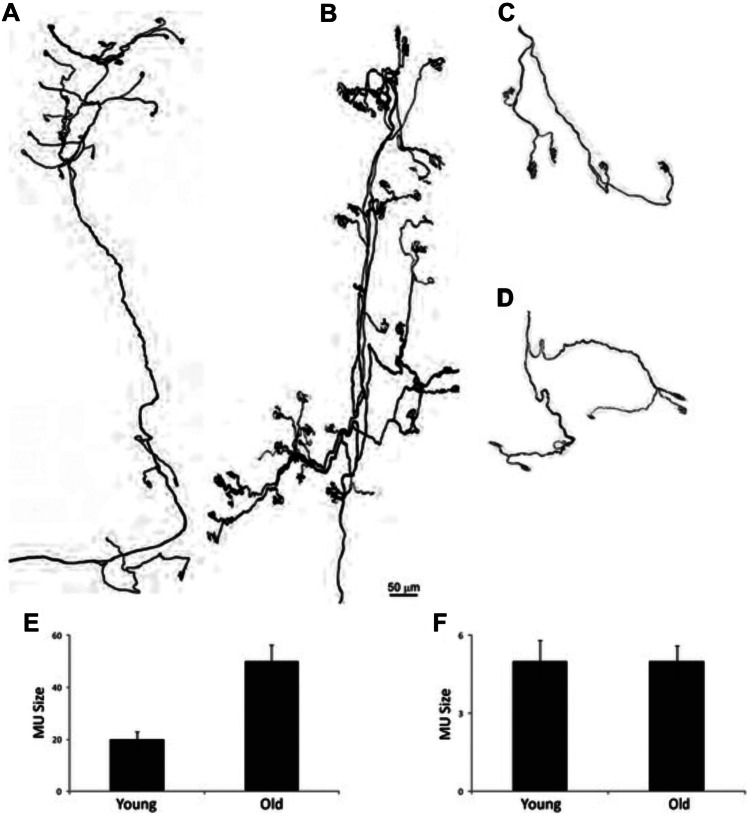
- Fischer LR, Culver DG, Tennant P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
