

Profiling epigenetic age in single cells
- PMID: 36211119
- PMCID: PMC9536112
- DOI: 10.1038/s43587-021-00134-3
Profiling epigenetic age in single cells
Abstract
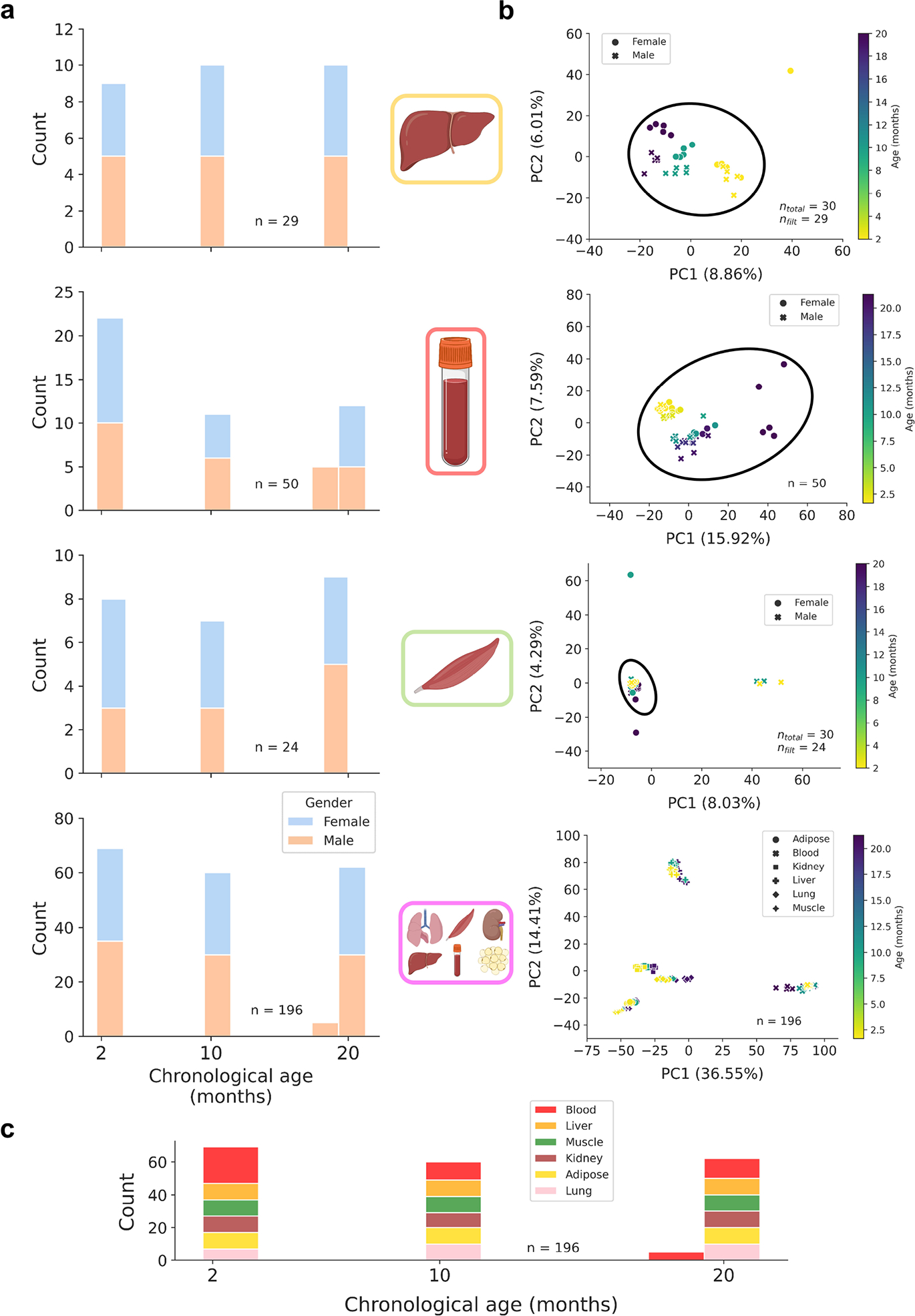
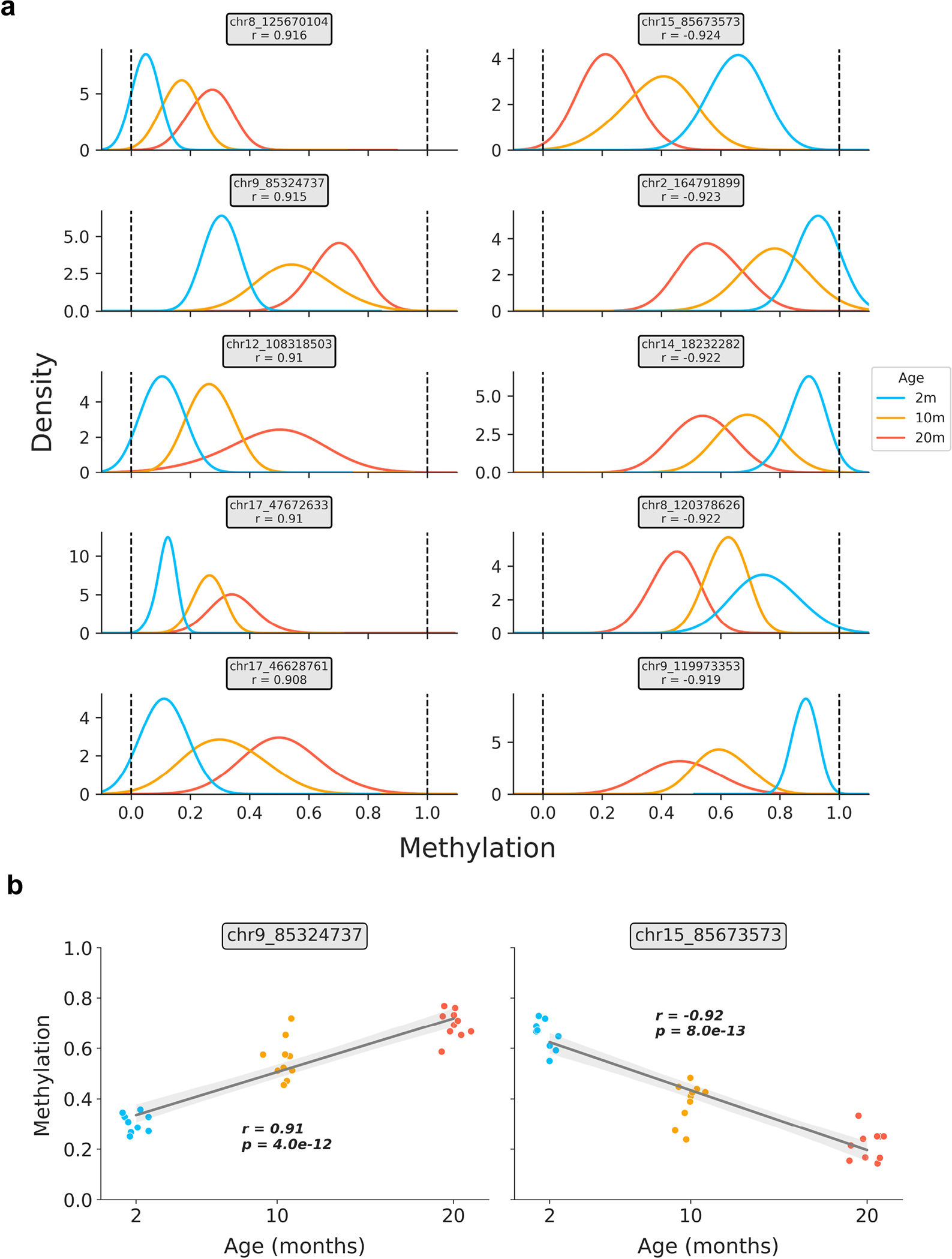
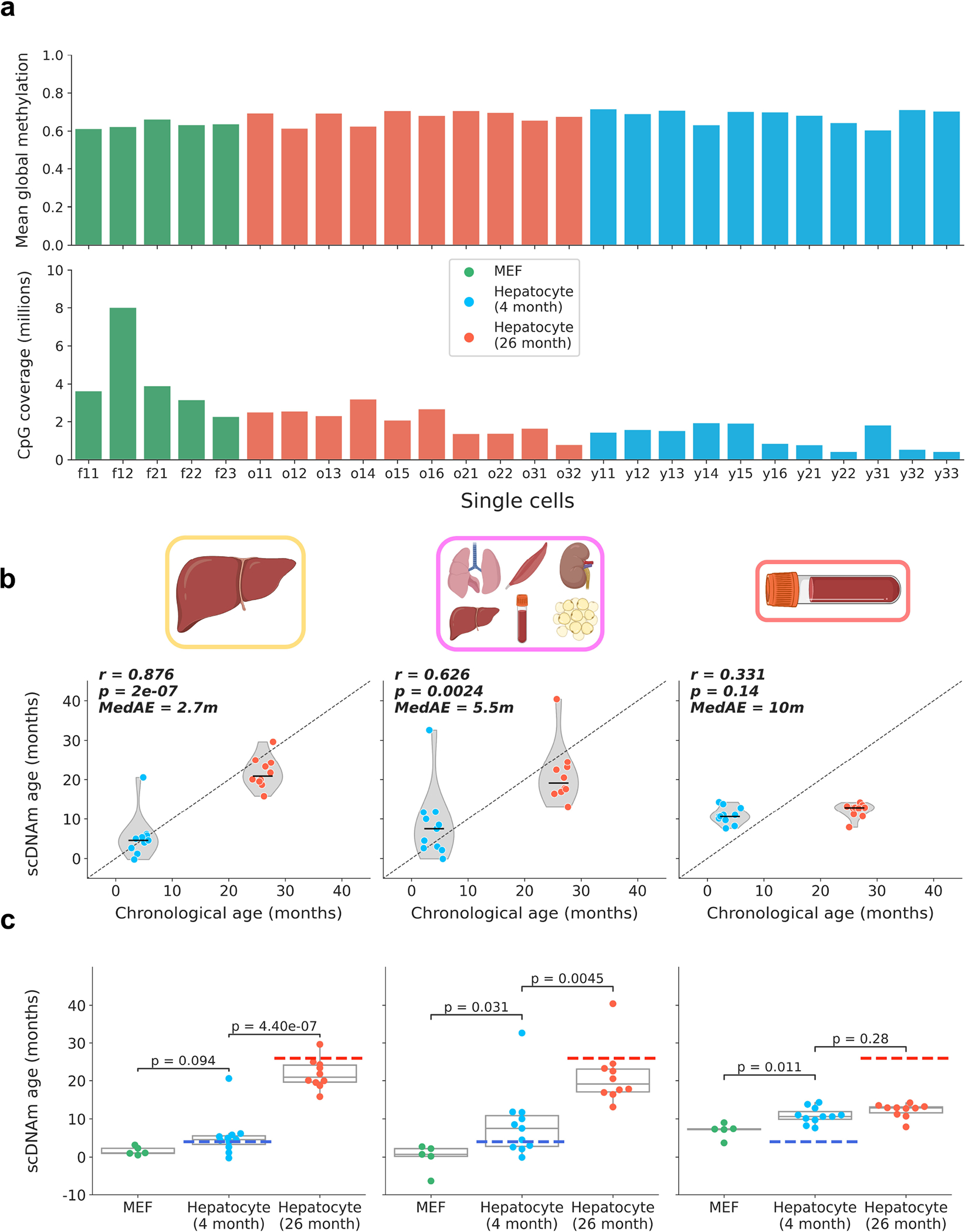
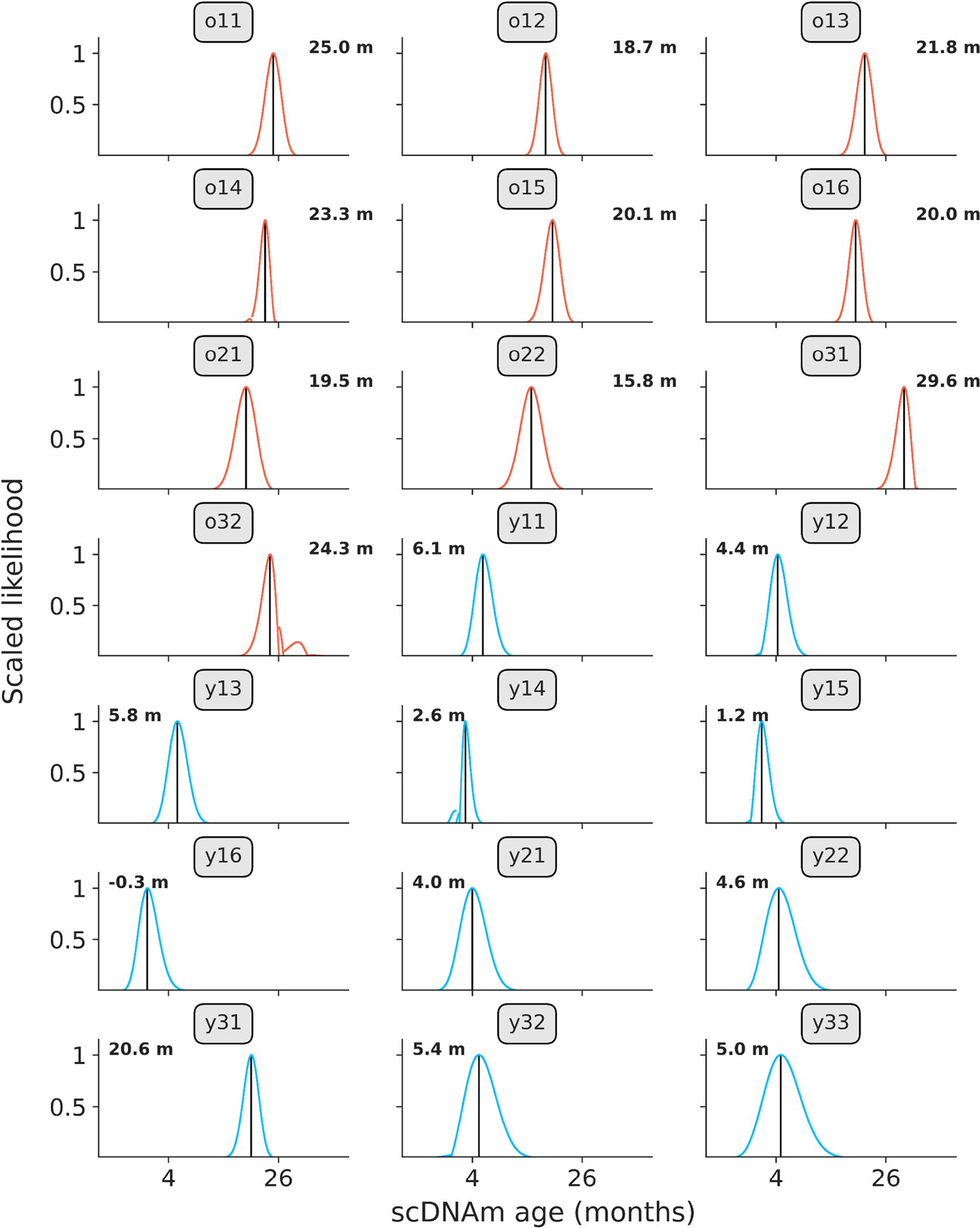
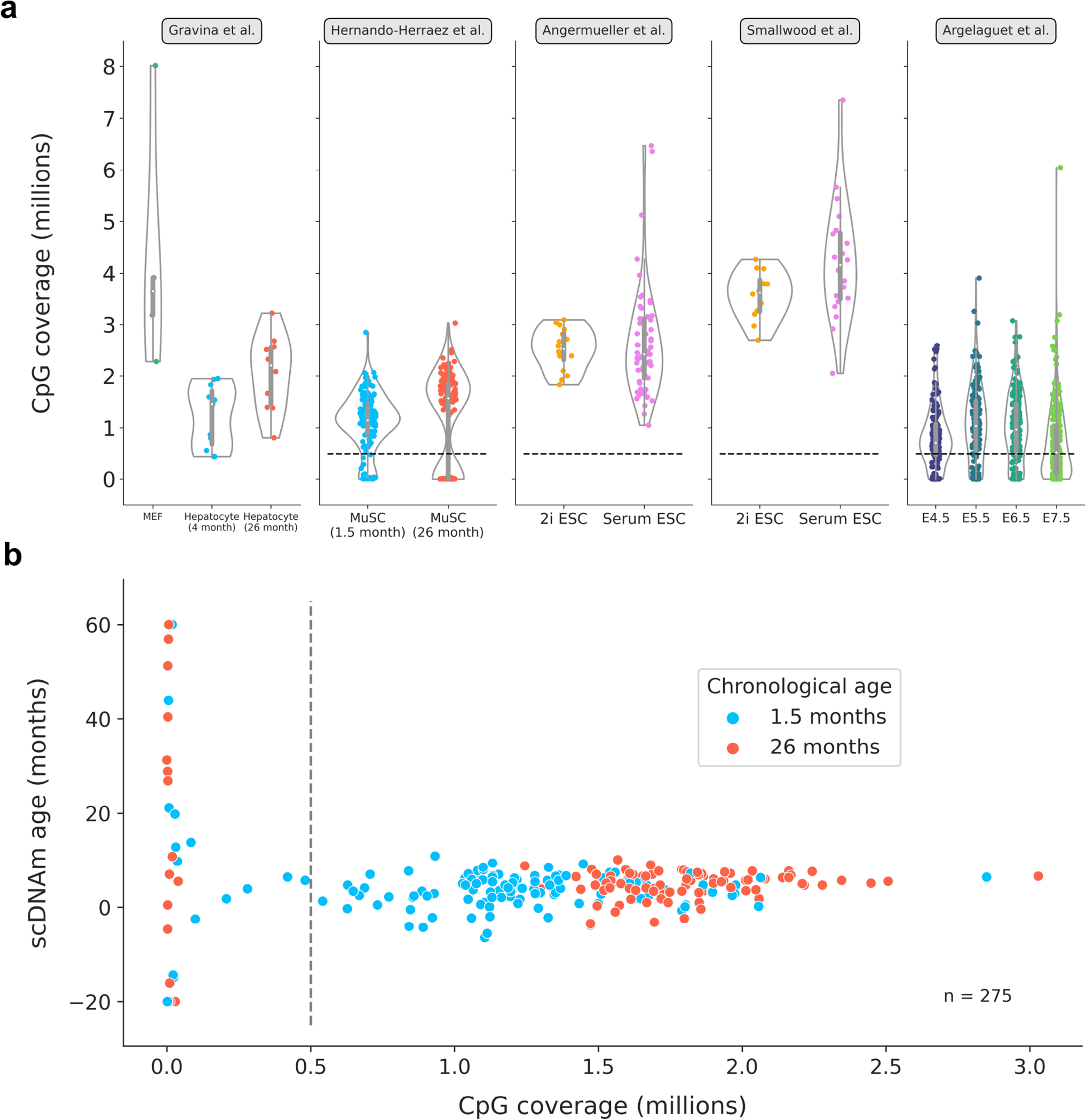
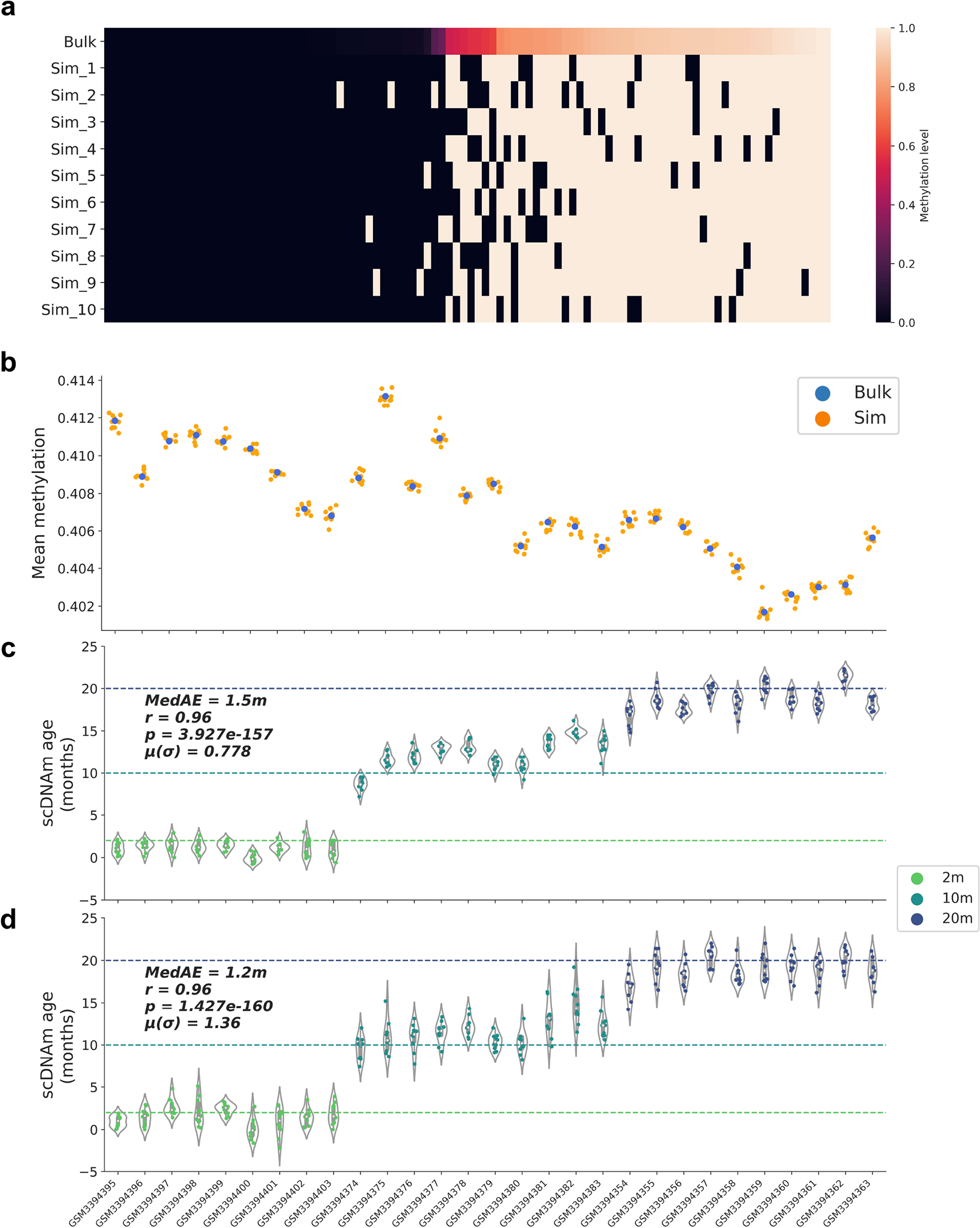
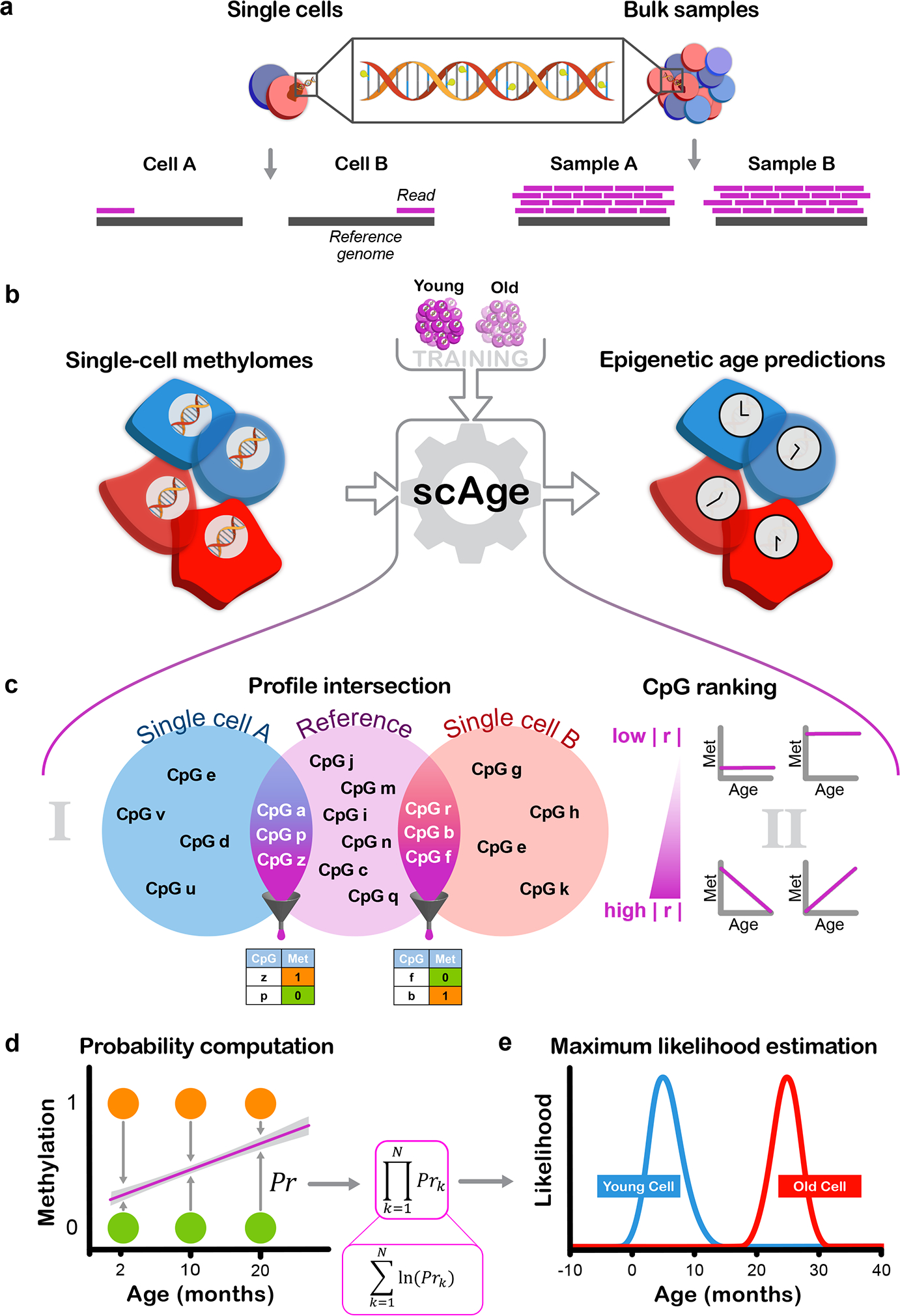
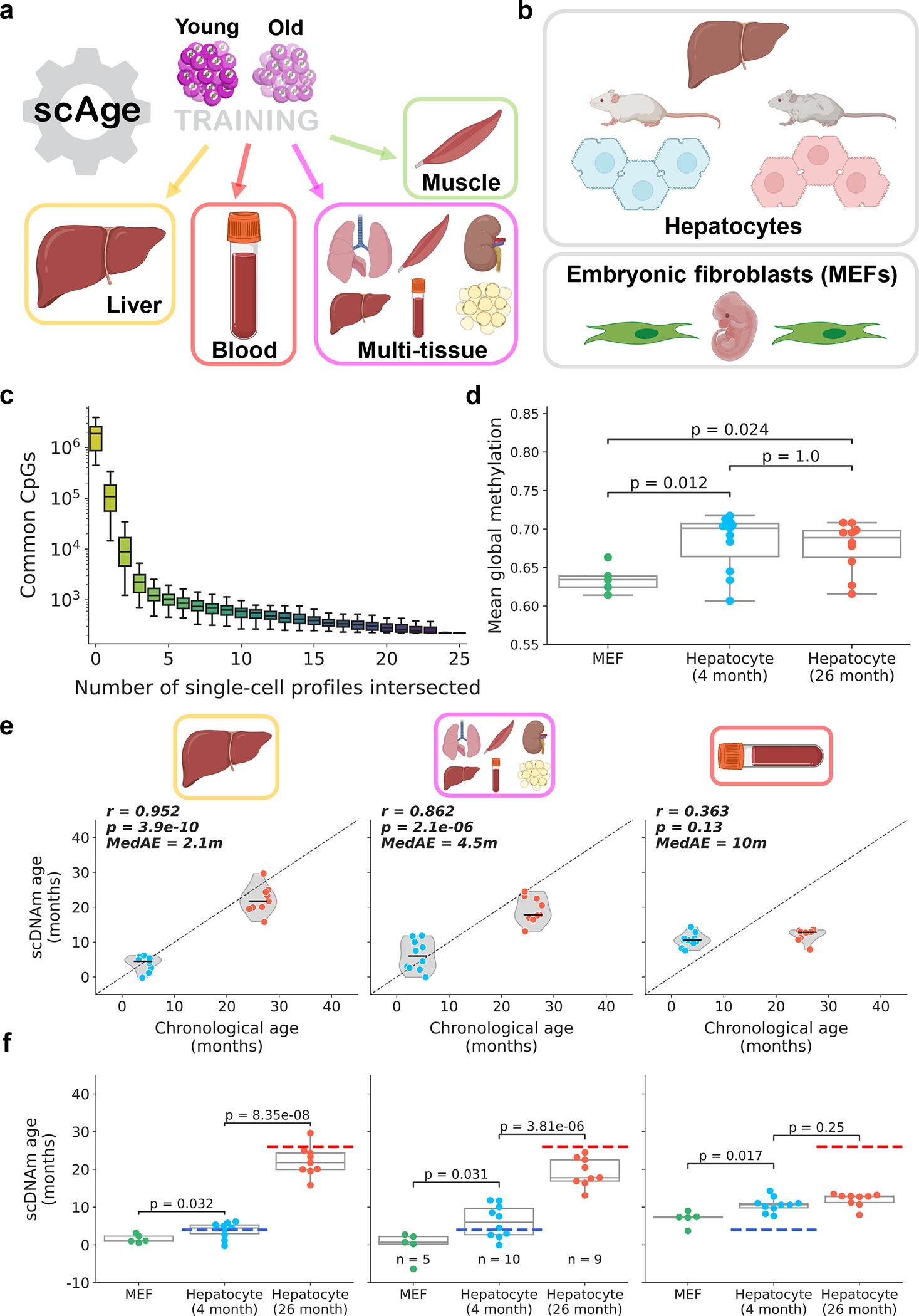
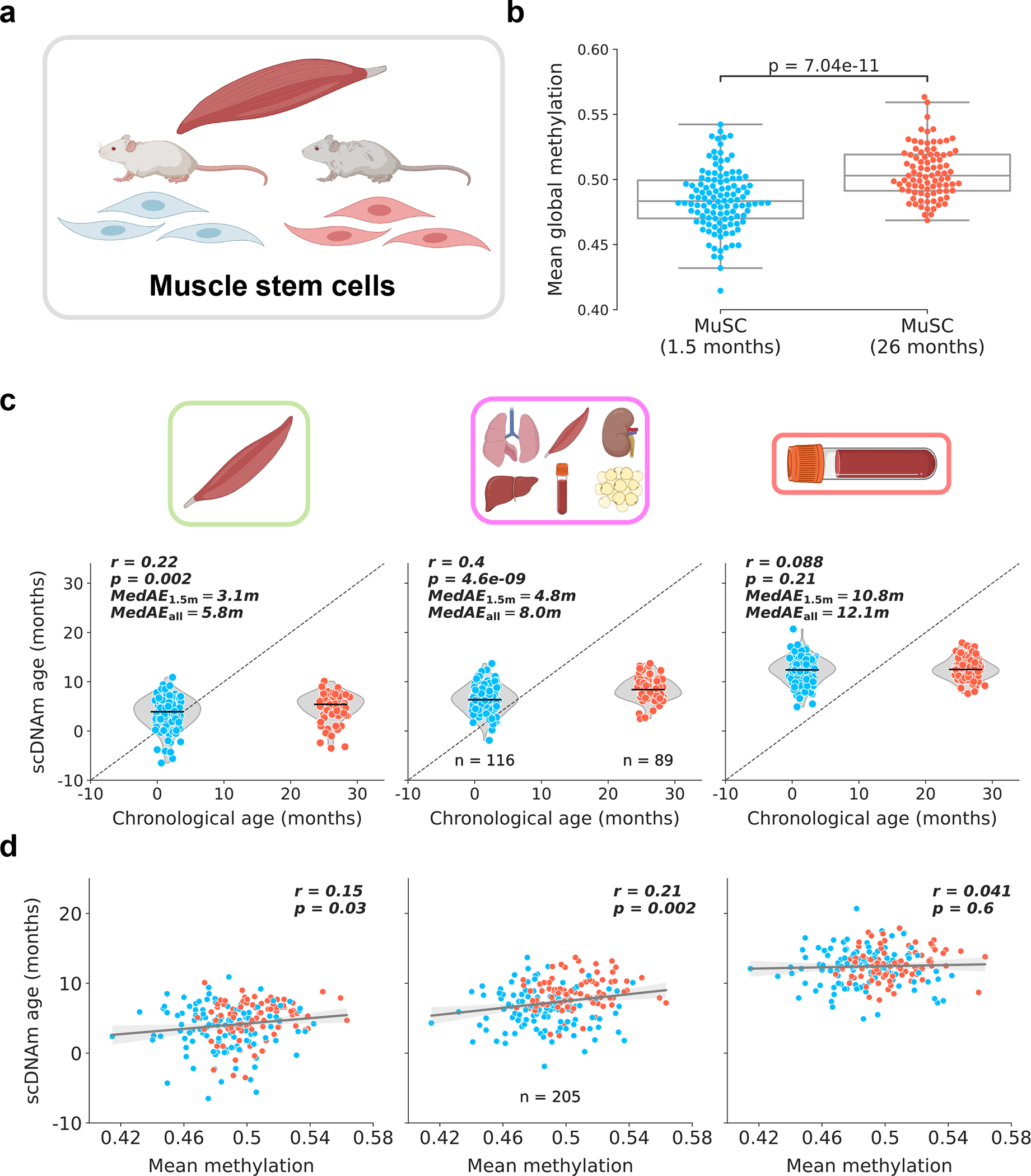
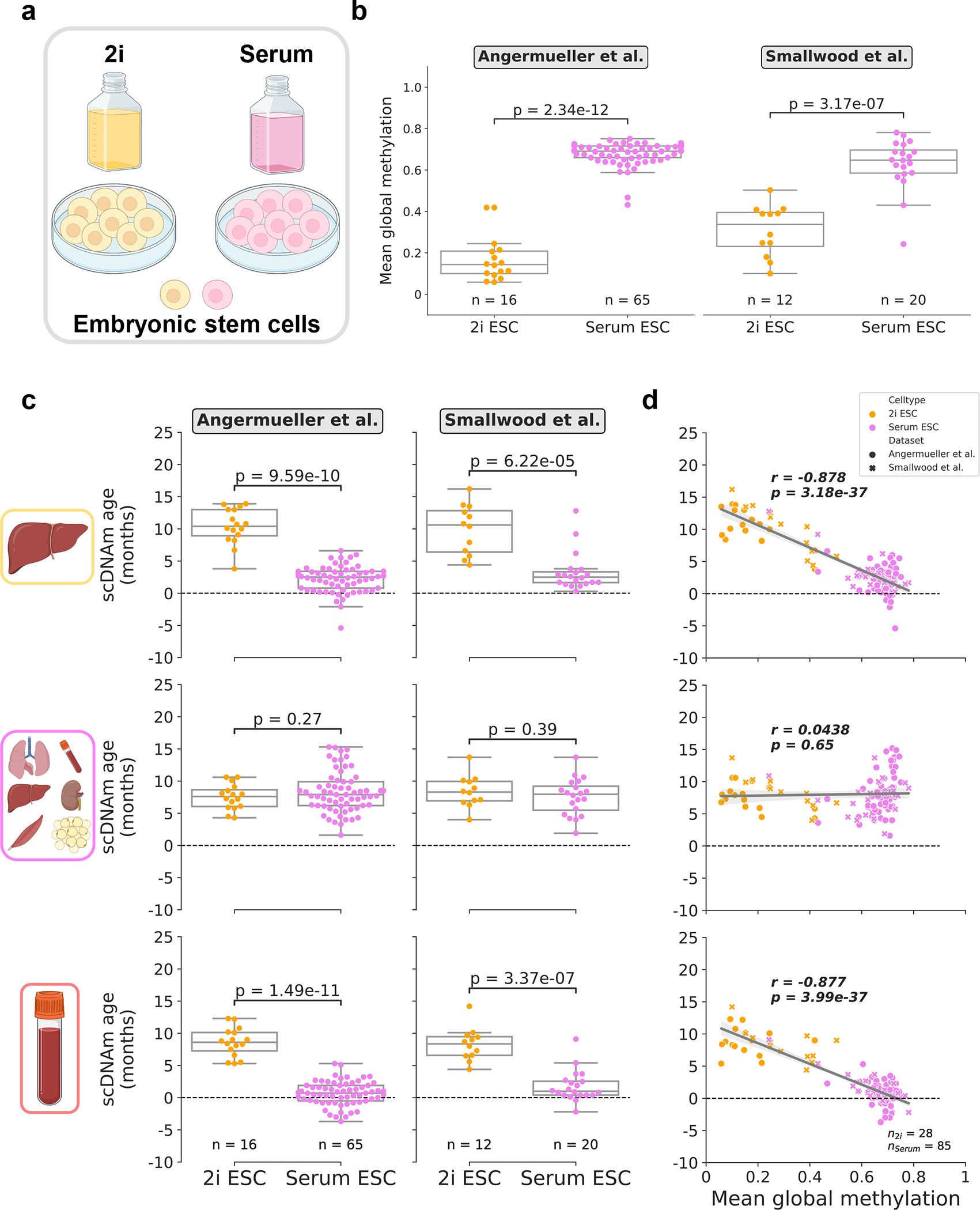
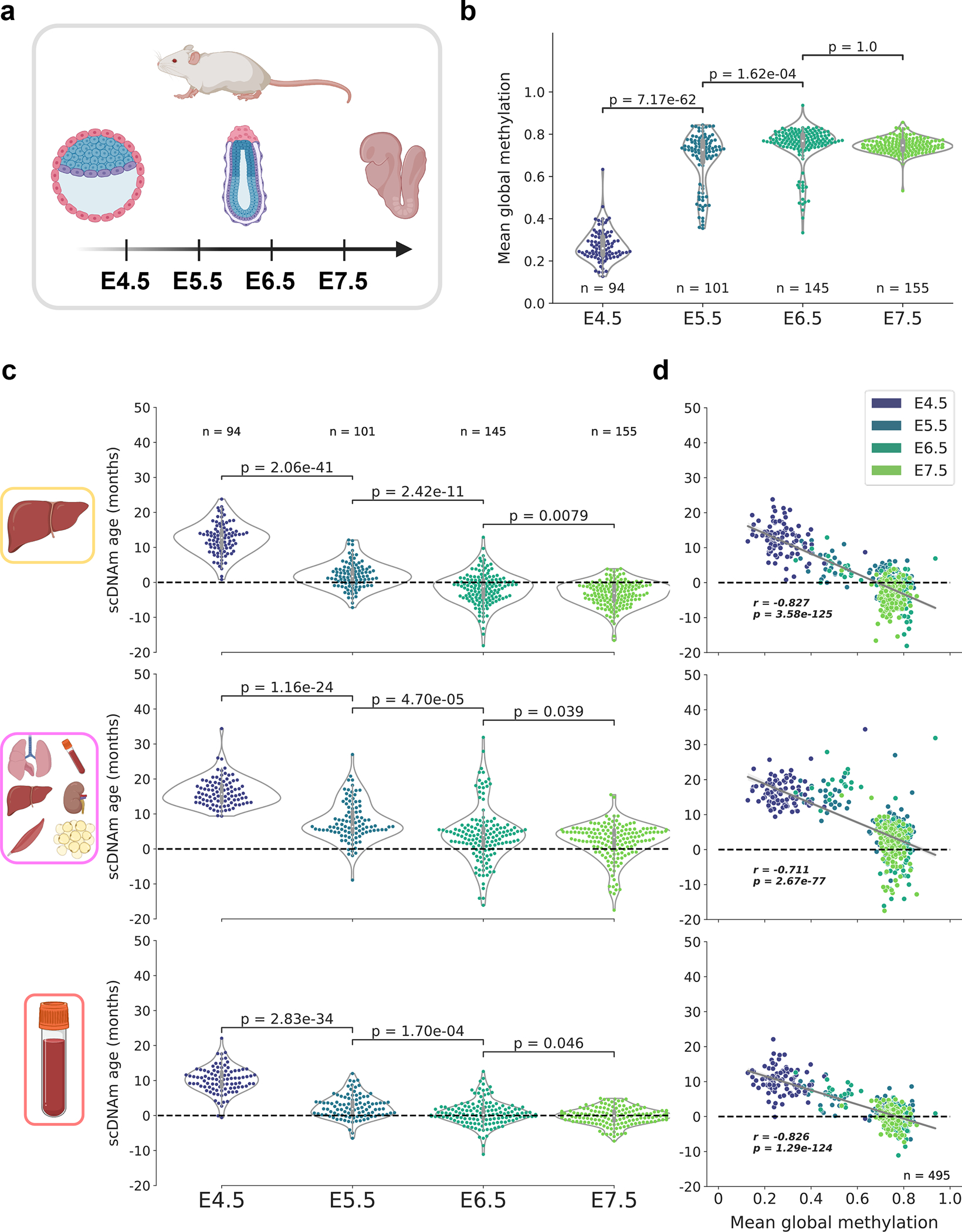
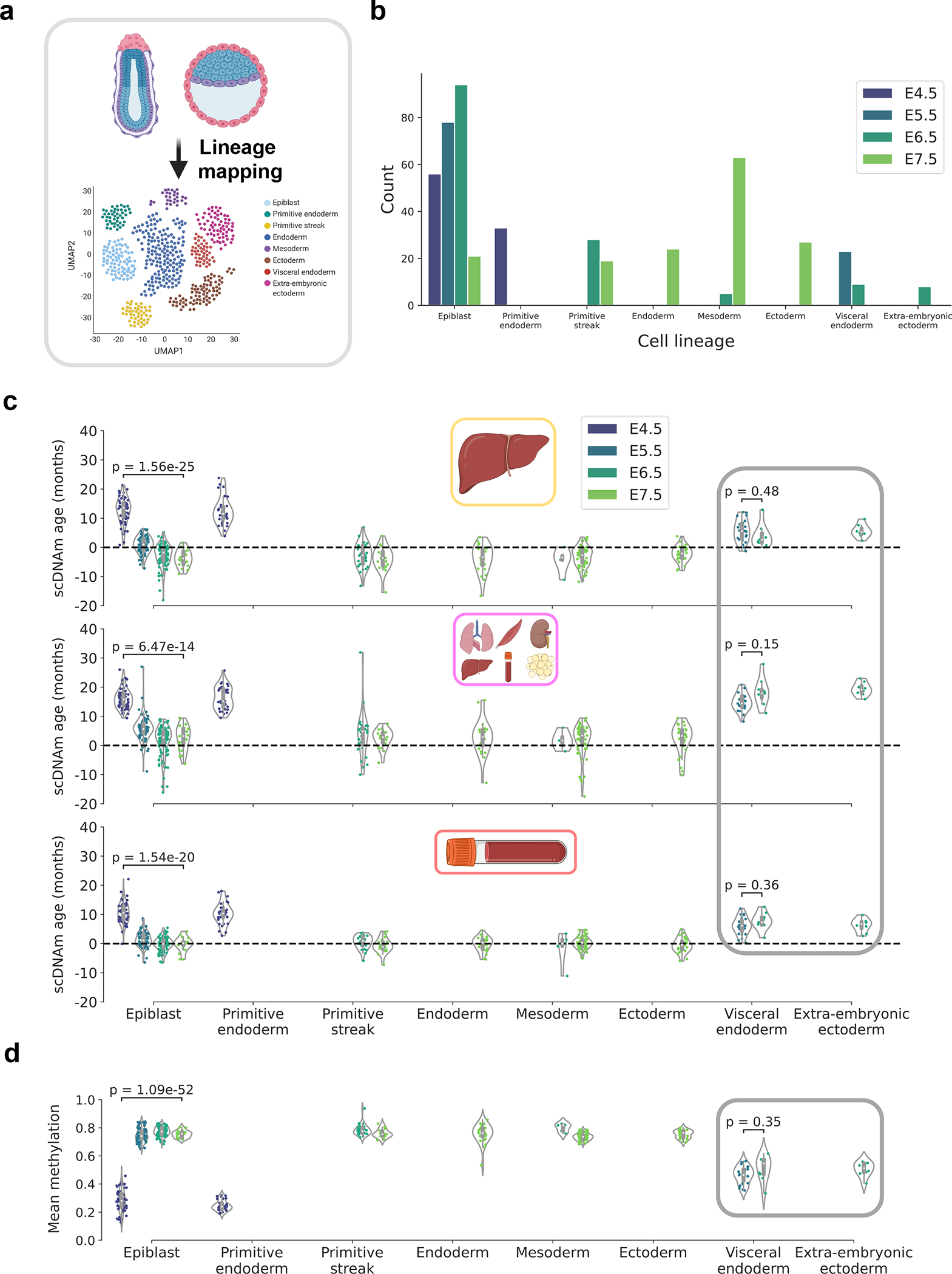
DNA methylation dynamics emerged as a promising biomarker of mammalian aging, with multivariate machine learning models ('epigenetic clocks') enabling measurement of biological age in bulk tissue samples. However, intrinsically sparse and binarized methylation profiles of individual cells have so far precluded the assessment of aging in single-cell data. Here, we introduce scAge, a statistical framework for epigenetic age profiling at single-cell resolution, and validate our approach in mice. Our method recapitulates the chronological age of tissues, while uncovering heterogeneity among cells. We show accurate tracking of the aging process in hepatocytes, demonstrate attenuated epigenetic aging in muscle stem cells, and track age dynamics in embryonic stem cells. We also use scAge to reveal, at the single-cell level, a natural and stratified rejuvenation event occurring during early embryogenesis. We provide our framework as a resource to enable exploration of epigenetic aging trajectories at single-cell resolution.
Conflict of interest statement
COMPETING INTERESTS Brigham and Women’s Hospital is the sole owner of a provisional patent application directed at this invention in which all authors, Alexandre Trapp, Csaba Kerepesi, and Vadim N. Gladyshev, are named inventors.
Figures
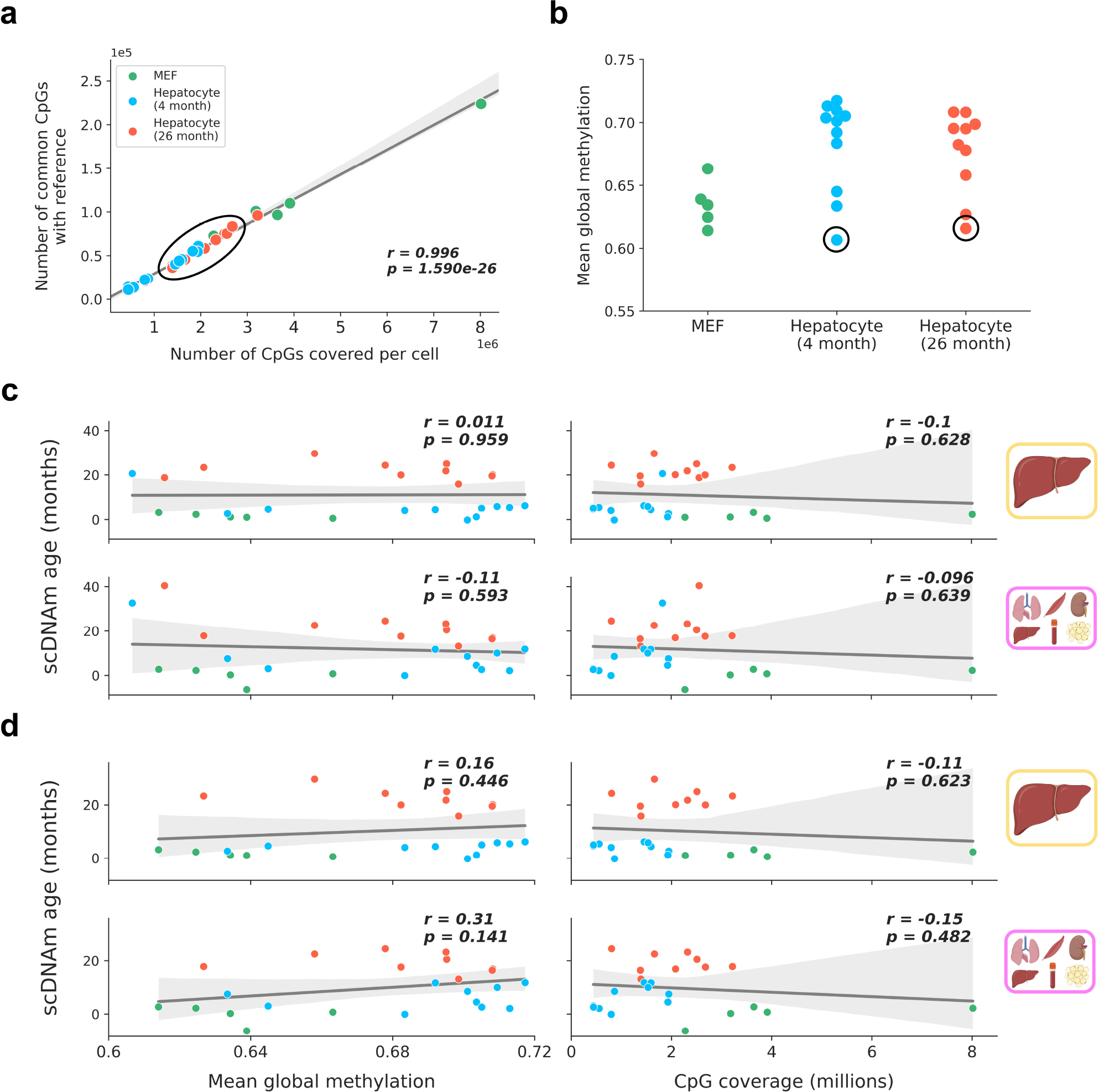
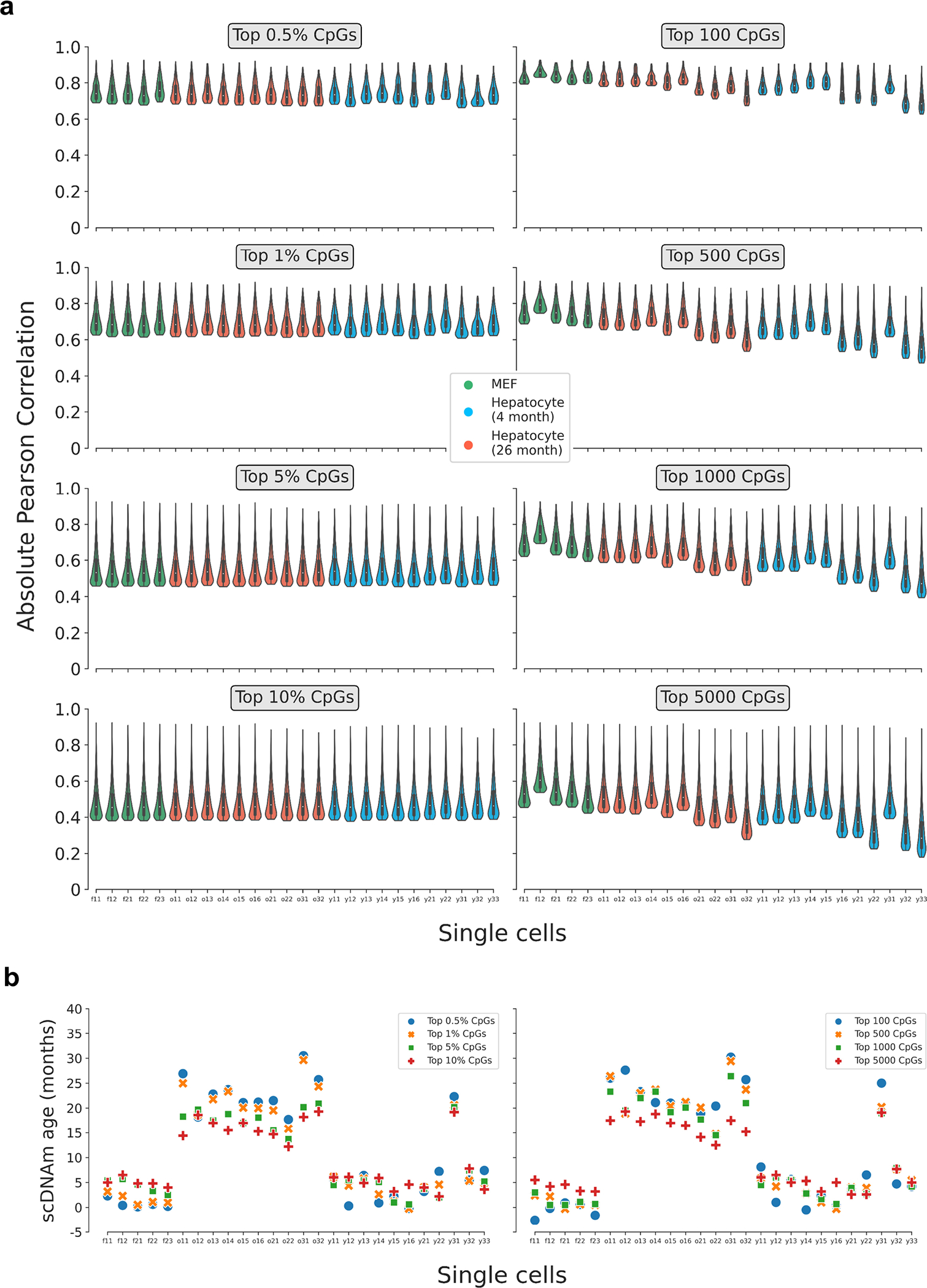
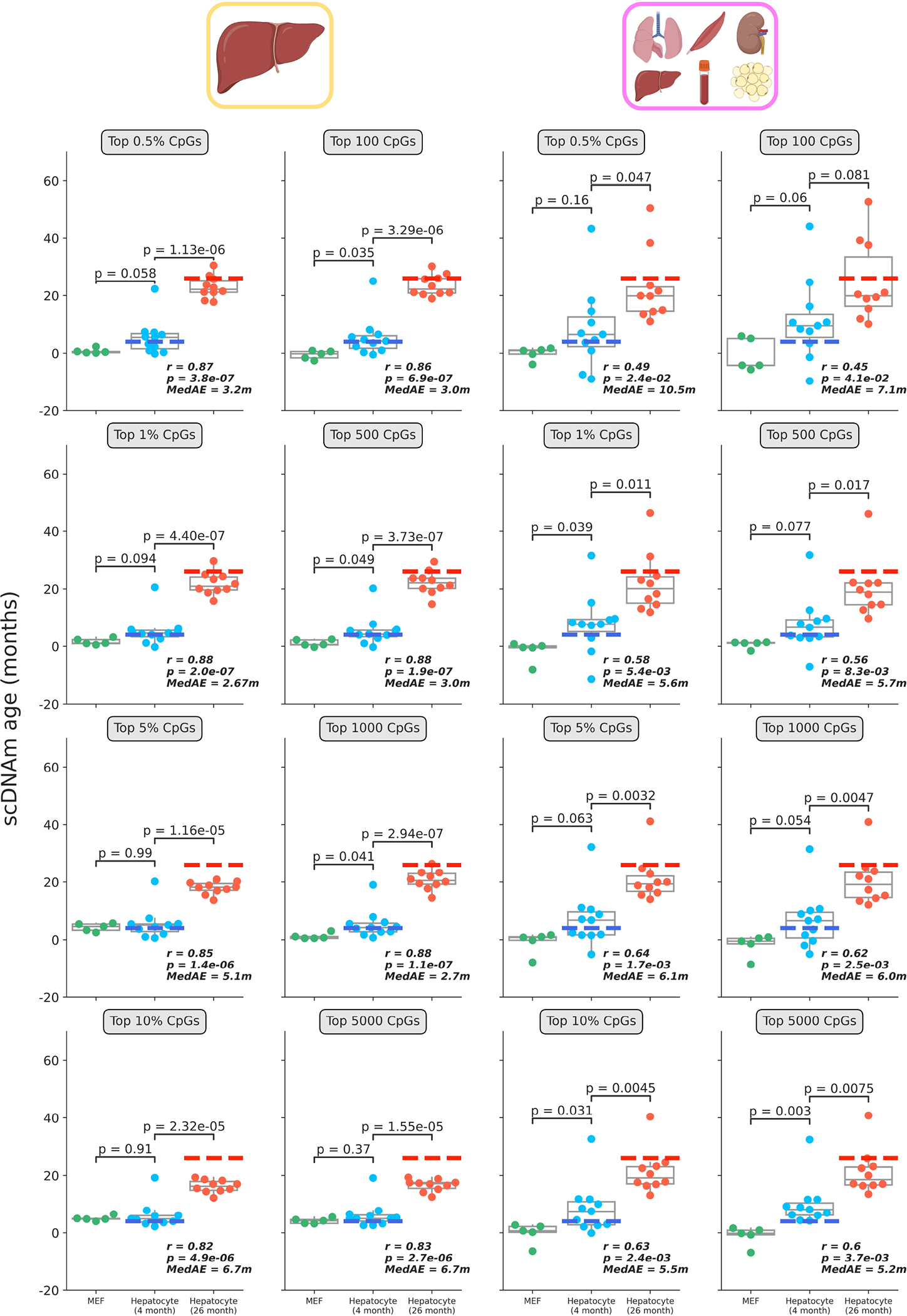
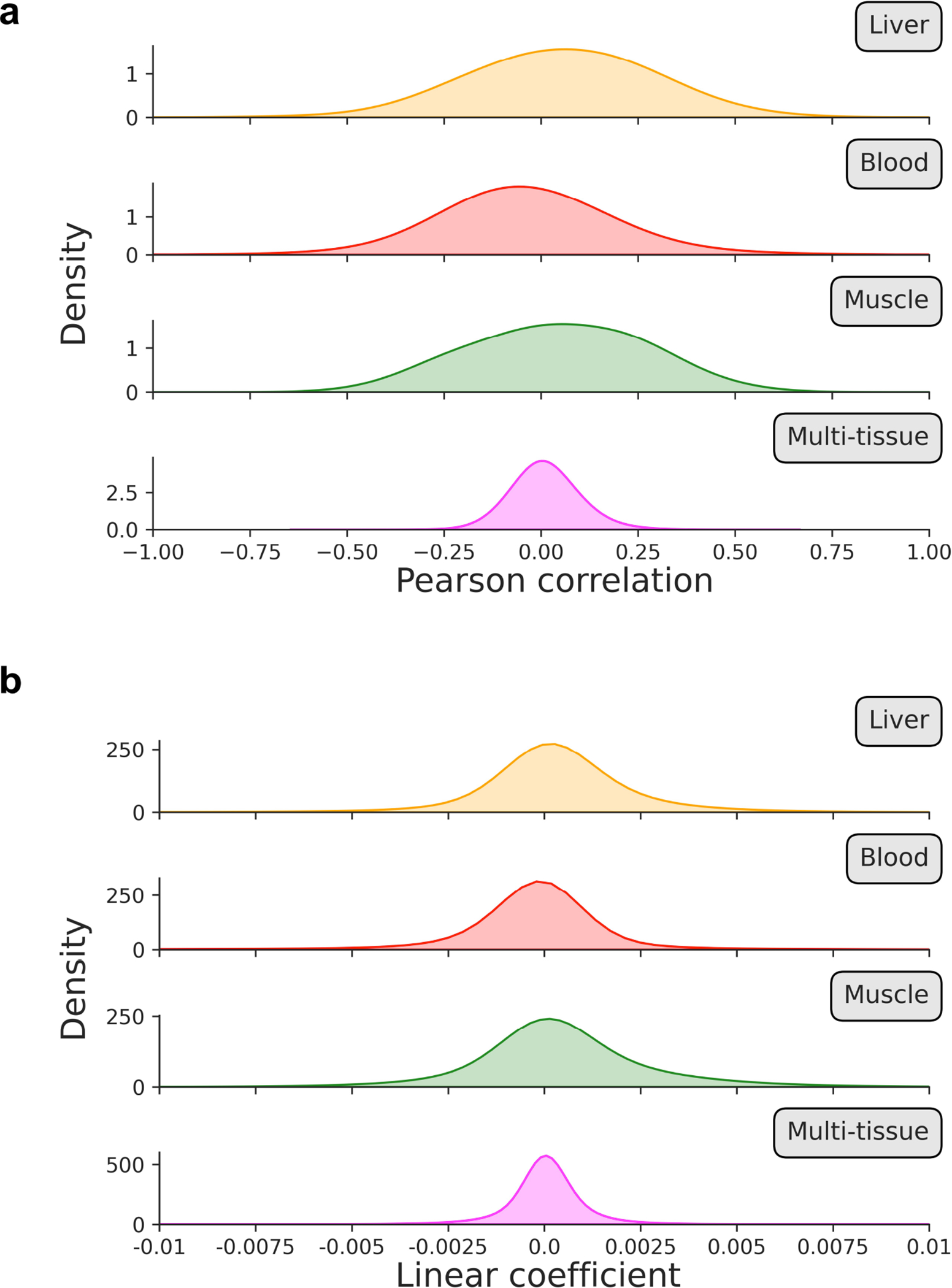
Comment in
-
DNA-methylation aging at single-cell level.Nat Aging. 2021 Dec;1(12):1086-1087. doi: 10.1038/s43587-021-00154-z. Nat Aging. 2021. PMID: 37117522 No abstract available.
Similar articles
-
Cell-type specific epigenetic clocks to quantify biological age at cell-type resolution.Aging (Albany NY). 2024 Dec 29;16(22):13452-13504. doi: 10.18632/aging.206184. Epub 2024 Dec 29. Aging (Albany NY). 2024. PMID: 39760516 Free PMC article.
-
Dysfunctional epigenetic aging of the normal colon and colorectal cancer risk.Clin Epigenetics. 2020 Jan 3;12(1):5. doi: 10.1186/s13148-019-0801-3. Clin Epigenetics. 2020. PMID: 31900199 Free PMC article.
-
Epigenetic clock: A promising biomarker and practical tool in aging.Ageing Res Rev. 2022 Nov;81:101743. doi: 10.1016/j.arr.2022.101743. Epub 2022 Oct 4. Ageing Res Rev. 2022. PMID: 36206857 Review.
-
TIME-seq reduces time and cost of DNA methylation measurement for epigenetic clock construction.Nat Aging. 2024 Feb;4(2):261-274. doi: 10.1038/s43587-023-00555-2. Epub 2024 Jan 10. Nat Aging. 2024. PMID: 38200273 Free PMC article.
-
DNA Methylation as a Biomarker of Aging in Epidemiologic Studies.Methods Mol Biol. 2018;1856:219-231. doi: 10.1007/978-1-4939-8751-1_12. Methods Mol Biol. 2018. PMID: 30178254 Review.
Cited by
-
DNA methylation in mammalian development and disease.Nat Rev Genet. 2025 Jan;26(1):7-30. doi: 10.1038/s41576-024-00760-8. Epub 2024 Aug 12. Nat Rev Genet. 2025. PMID: 39134824 Review.
-
CellBiAge: Improved single-cell age classification using data binarization.Cell Rep. 2023 Dec 26;42(12):113500. doi: 10.1016/j.celrep.2023.113500. Epub 2023 Nov 30. Cell Rep. 2023. PMID: 38032797 Free PMC article.
-
The Cutting Edge of Epigenetic Clocks: In Search of Mechanisms Linking Aging and Mental Health.Biol Psychiatry. 2023 Nov 1;94(9):694-705. doi: 10.1016/j.biopsych.2023.02.001. Epub 2023 Feb 9. Biol Psychiatry. 2023. PMID: 36764569 Free PMC article. Review.
-
How is Big Data reshaping preclinical aging research?Lab Anim (NY). 2023 Dec;52(12):289-314. doi: 10.1038/s41684-023-01286-y. Epub 2023 Nov 28. Lab Anim (NY). 2023. PMID: 38017182 Review.
-
Application of a mathematical model to clarify the statistical characteristics of a pan-tissue DNA methylation clock.Geroscience. 2024 Apr;46(2):2001-2015. doi: 10.1007/s11357-023-00949-5. Epub 2023 Oct 3. Geroscience. 2024. PMID: 37787856 Free PMC article.
References
-
- Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
